Whole genome sequencing and evolutionary analysis of bovine ephemeral fever virus isolate JM 2020 in Guangdong Province
-
摘要:目的
分析广东省牛流行热病毒(Bovine ephemeral fever virus,BEFV)分离株JM 2020与其他地区毒株的进化关系,阐明遗传进化与全基因组的特征,为中国乃至世界对牛流行热疾病的流行情况以及预防该疾病提供有用信息。
方法根据从GenBank下载的BEFV毒株的全基因组序列信息,设计引物PCR扩增糖蛋白(Glycoprotein,G)基因;另设计10对引物用于扩增全基因组,送测序得到10个片段的基因序列,运用DNAStar软件中的EditSeq手动编辑和拼接获得全基因组序列。运用MEGA 6.0分别构建G基因和全基因组进化树进行进化分析。
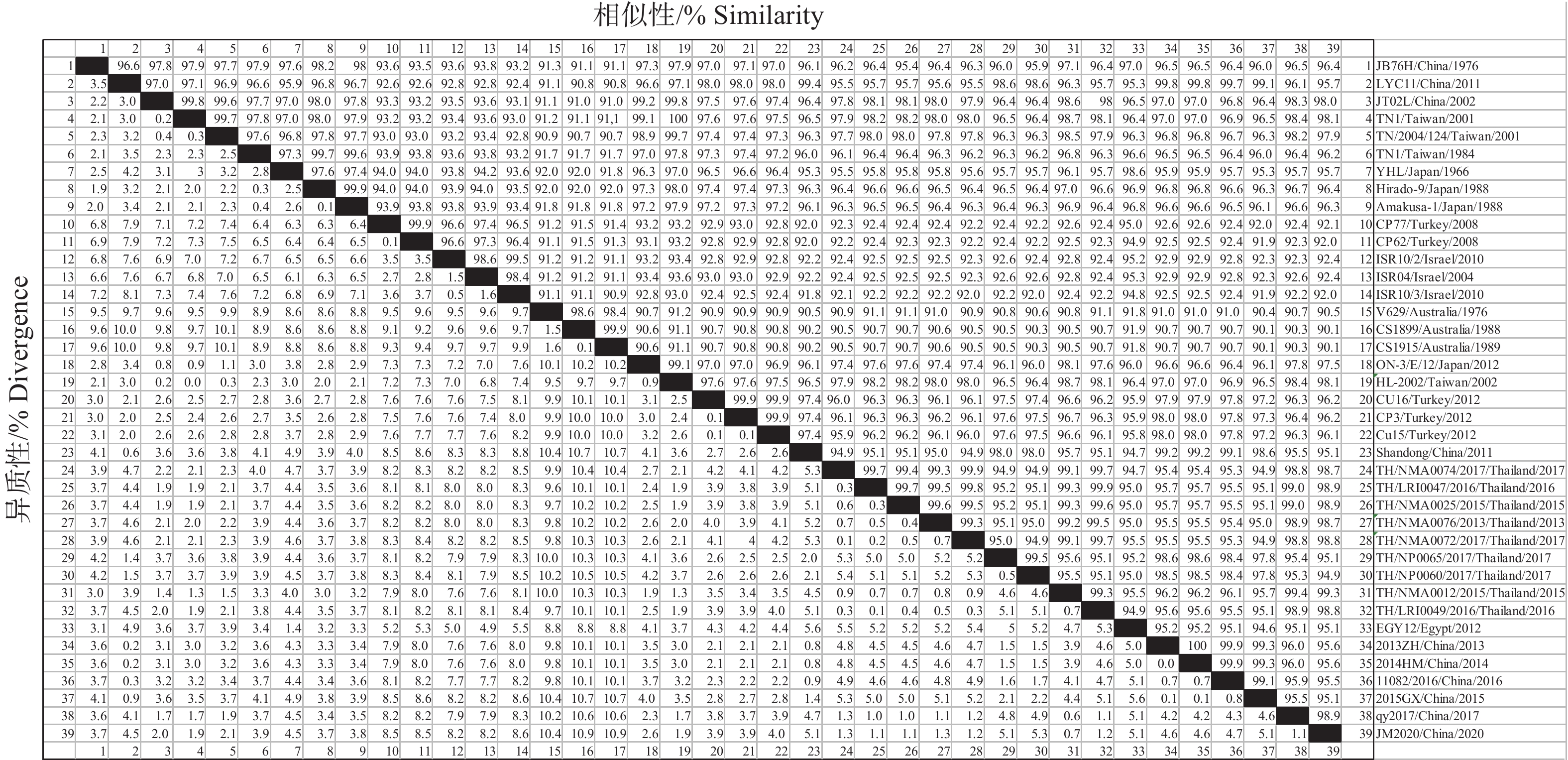
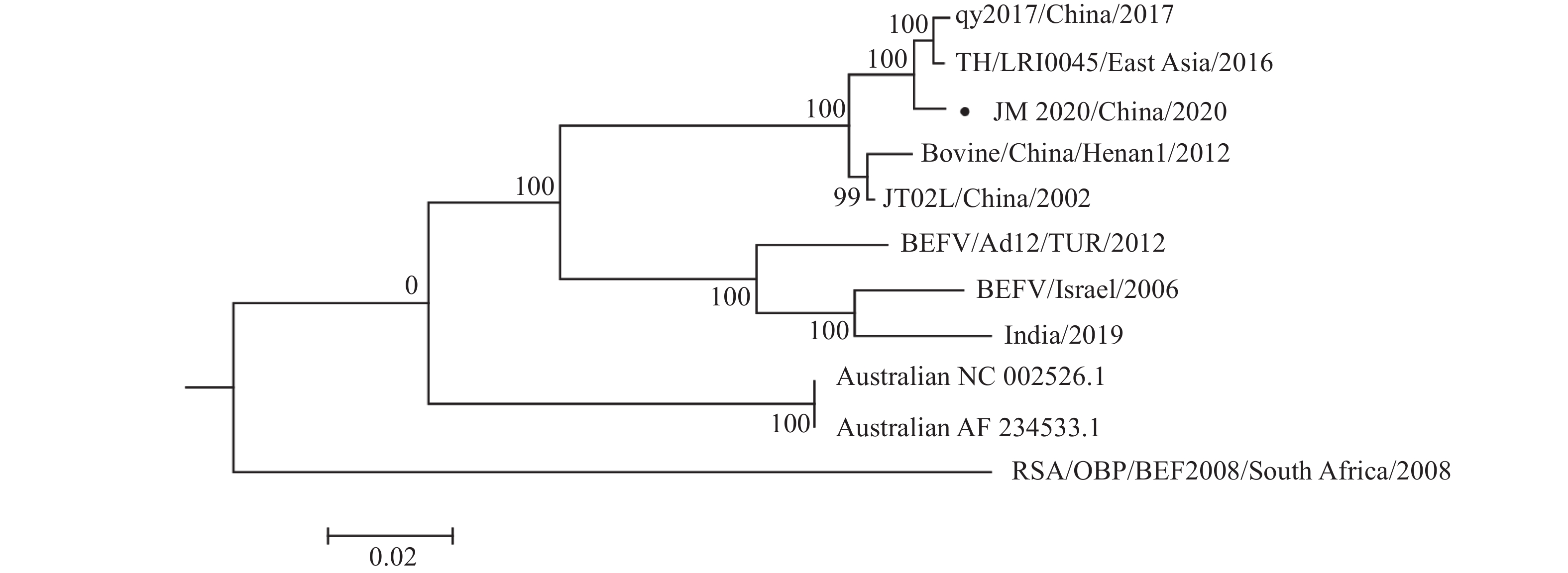
结果JM 2020毒株的G基因和全基因组与泰国毒株的核苷酸序列相似性均最高,分别为94.9%~99.3%和99.0%,进化分析表明JM 2020与2013—2017年泰国毒株处于同一小分支,而与JB76H、JT02L等国内毒株有一定进化距离。全基因组测序结果表明,JM 2020全基因组长度为14 902个核苷酸(nt),包括50 nt的前导序列,1 296 nt的核蛋白(Nucleoprotein,N)基因,837 nt的磷蛋白(Phosphoprotein,P)基因,672 nt的基质蛋白(Matrixprotein,M)基因,1 872 nt的G基因,1 812 nt的非结构糖蛋白II (Non-structural glycoprotein II,GNS)基因,618 nt的α1、α2基因,444 nt的β基因,345 nt的γ基因,444 nt的RNA依赖性聚合酶(Large multi-functional enzyme,L)基因和73 nt的尾随序列,且分别被21、47、68、67、23、64、59、79和35 nt的区域隔开。JM 2020、qy2017及泰国毒株均出现P′基因(P基因多顺反子产物)截断的特征。
结论JM 2020毒株与2017年分离毒株qy2017序列相似性高且均与泰国毒株进化关系较近,与JB76H等我国其他地区毒株有一定进化距离。该研究丰富了中国BEFV流行株的基因组信息,为牛流行热疫病综合防控及开发新疫苗奠定了基础。
Abstract:ObjectiveThe purpose of this study was to analyze the evolutionary relationship between bovine ephemeral fever virus (BEFV) isolate JM 2020 in Guangdong Province and other strains from other regions, and clarify the characteristics of genetic evolution and whole genome, so as to provide information for the epidemic situation and prevention of bovine ephemeral fever disease in China and the world.
MethodAccording to the whole genome sequence information of BEFV strain downloaded from GenBank, primers were designed to amplify glycoprotein (G) gene by PCR. The 10 pairs of primers were designed to amplify the whole genome, and the gene sequences of 10 fragments were obtained by sequencing. The whole genome sequences were manually edited and spliced by EditSeq in DNAStar software. Using MEGA 6.0 to construct G gene and whole genome evolutionary tree respectively for evolutionary analysis.
ResultThe G gene and whole genome of the JM 2020 strain had the highest nucleotide sequence similarity with the Thai strain, which were 94.9%−99.3% and 99.0% respectively. The evolutionary analysis showed that JM 2020 was in the same small branch with the 2013 to 2017 Thai strains, while there was a certain evolutionary distance from domestic virus strains such as JB76H and JT02L. Whole genome sequencing results showed that whole genome of JM 2020 was 14 902 nucleotides (nt) in length, including 50 nt leader sequence, 1 296 nt nucleoprotein (N) gene, and 837 nt phosphoprotein (P) gene, 672 nt matrixprotein (M) gene, 1 872 nt G gene, 1 812 nt non-structural glycoprotein II (GNS) gene, 618 nt α1 and α2 gene, 444 nt β gene, 345 nt γ gene, 6 444 nt large multi-functional enzyme (L) gene and tail sequence of 73 nt, which was separated by 21, 47, 68, 67, 23, 64, 59, 79 and 35 nt intergenic regions. The P′ gene (polycistronic product of P gene) of JM 2020 was truncated like qy2017 and the Thai strain.
ConclusionThe JM 2020 strain is highly similar with the qy2017 strain isolated in 2017 and they are closely related to the evolution of the Thai strain. They have some evolutionary distance with JB76H strain etc. from other regions of China. This study enriches the genome information of BEFV epidemic strains in China and lays a foundation for the prevention and control of bovine ephemeral fever and the research of new vaccines.
-
-
![]()
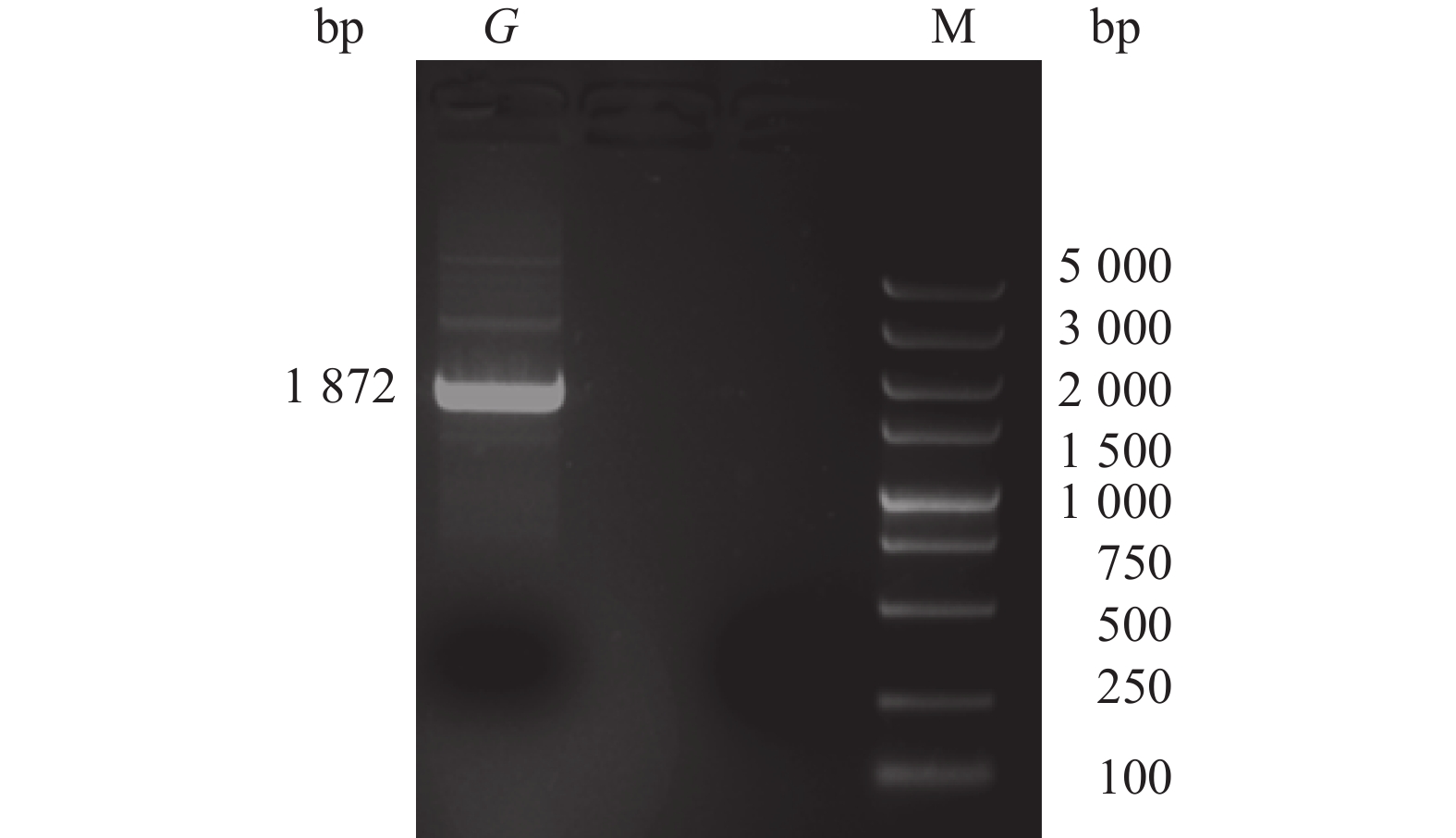
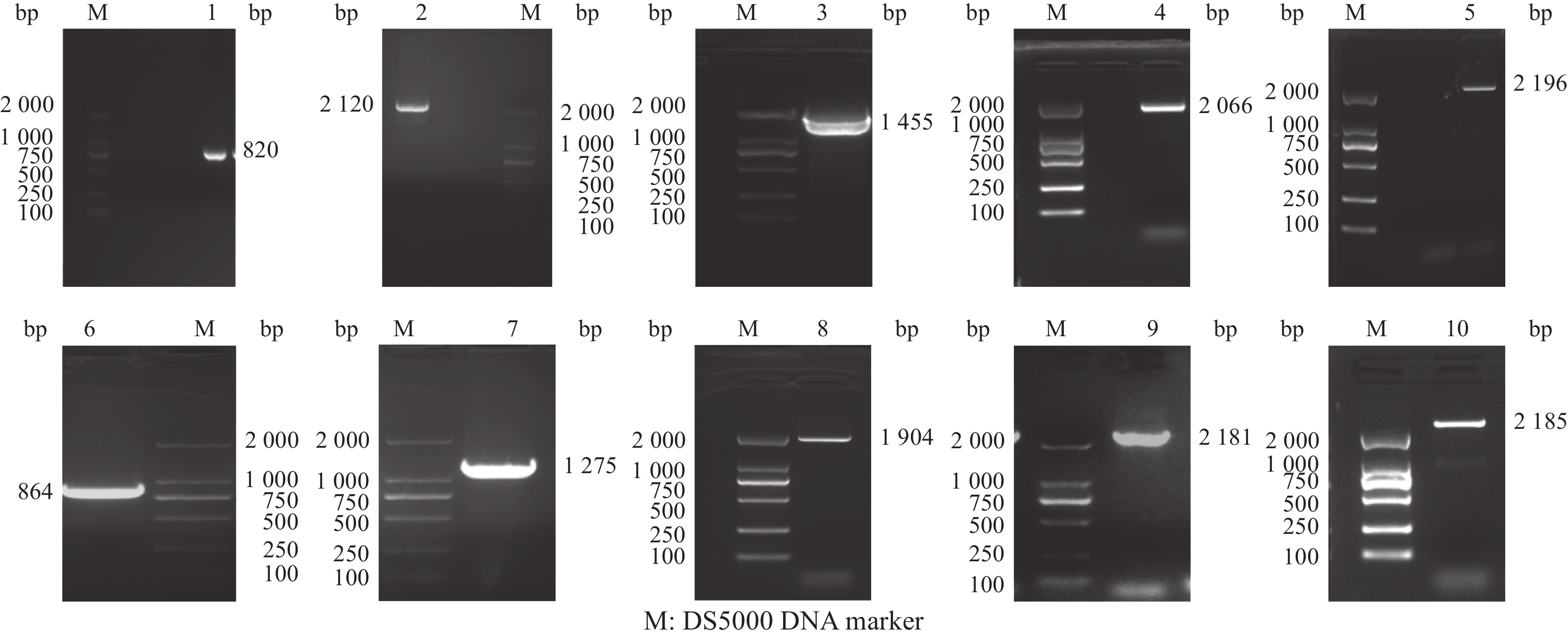
图 2 全基因组(G基因及10个片段)PCR扩增结果
Figure 2. Results of whole genome (G gene and 10 fragments) amplification by PCR
![]()
图 3 JM 2020 G基因进化树
“●”为JM 2020毒株;“○”为本研究检测到的毒株
Figure 3. Phylogenetic tree of JM 2020 G gene
“●” is the JM 2020 strain; “○” is the strain detected in this study
![]()
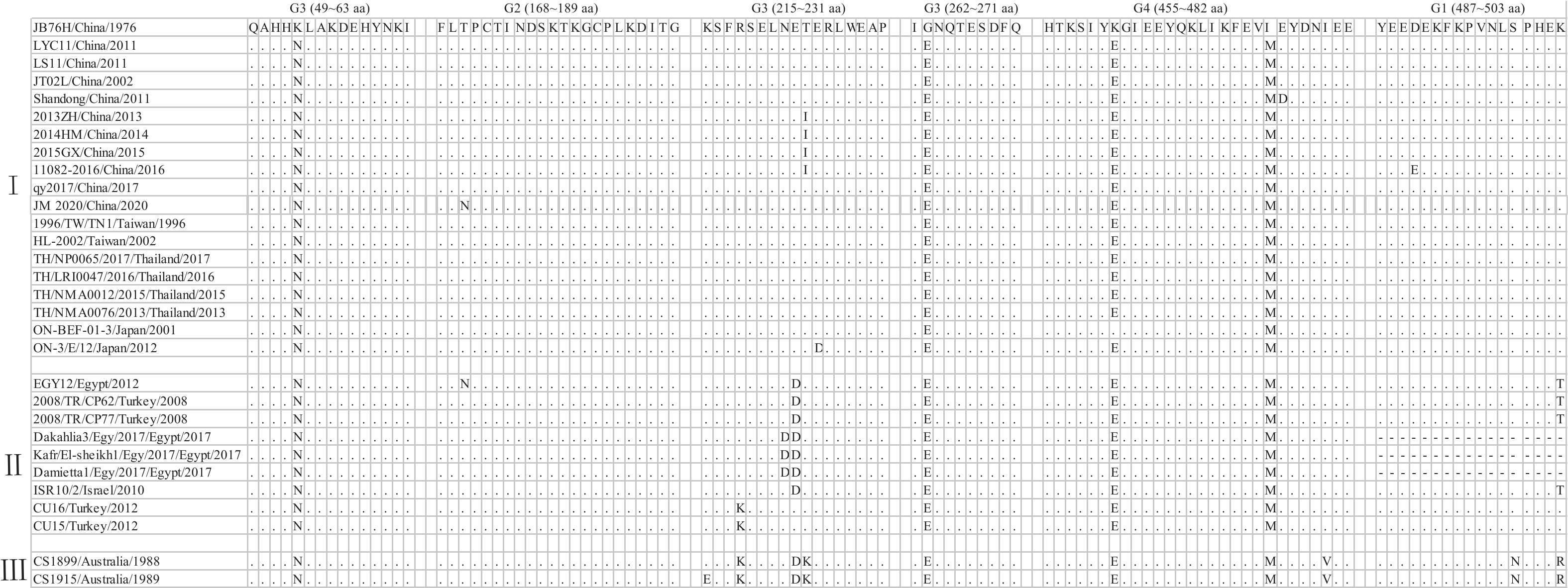
图 4 JM 2020毒株G基因核苷酸相似性分析(部分)
Figure 4. Nucleotide similarity analysis of G gene of JM 2020 strain (part)
![]()
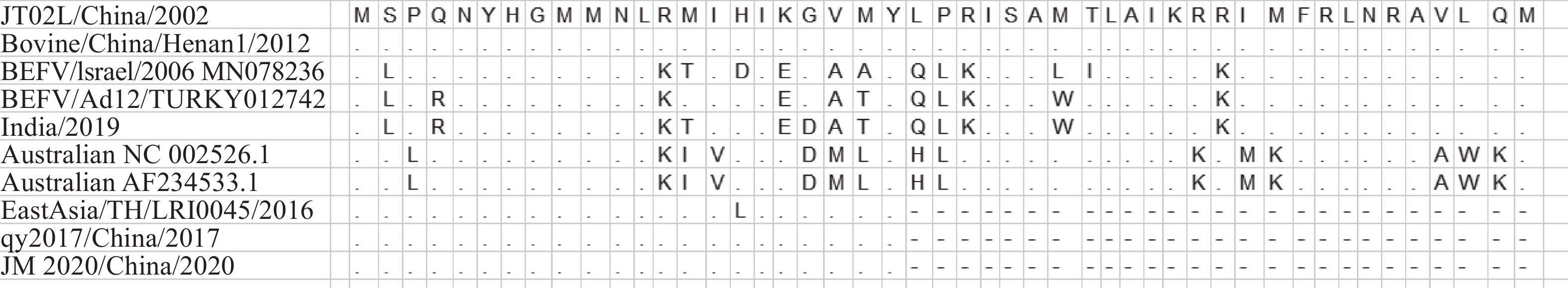
图 5 JM 2020毒株G蛋白抗原位点分析
“.”为与对照组毒株氨基酸相同;“-”为该位置氨基酸缺失
Figure 5. Antigenic sites of G protein of JM 2020 strain
“.” is the same amino acid as the control group; “-” is the amino acid deletion at this position
![]()
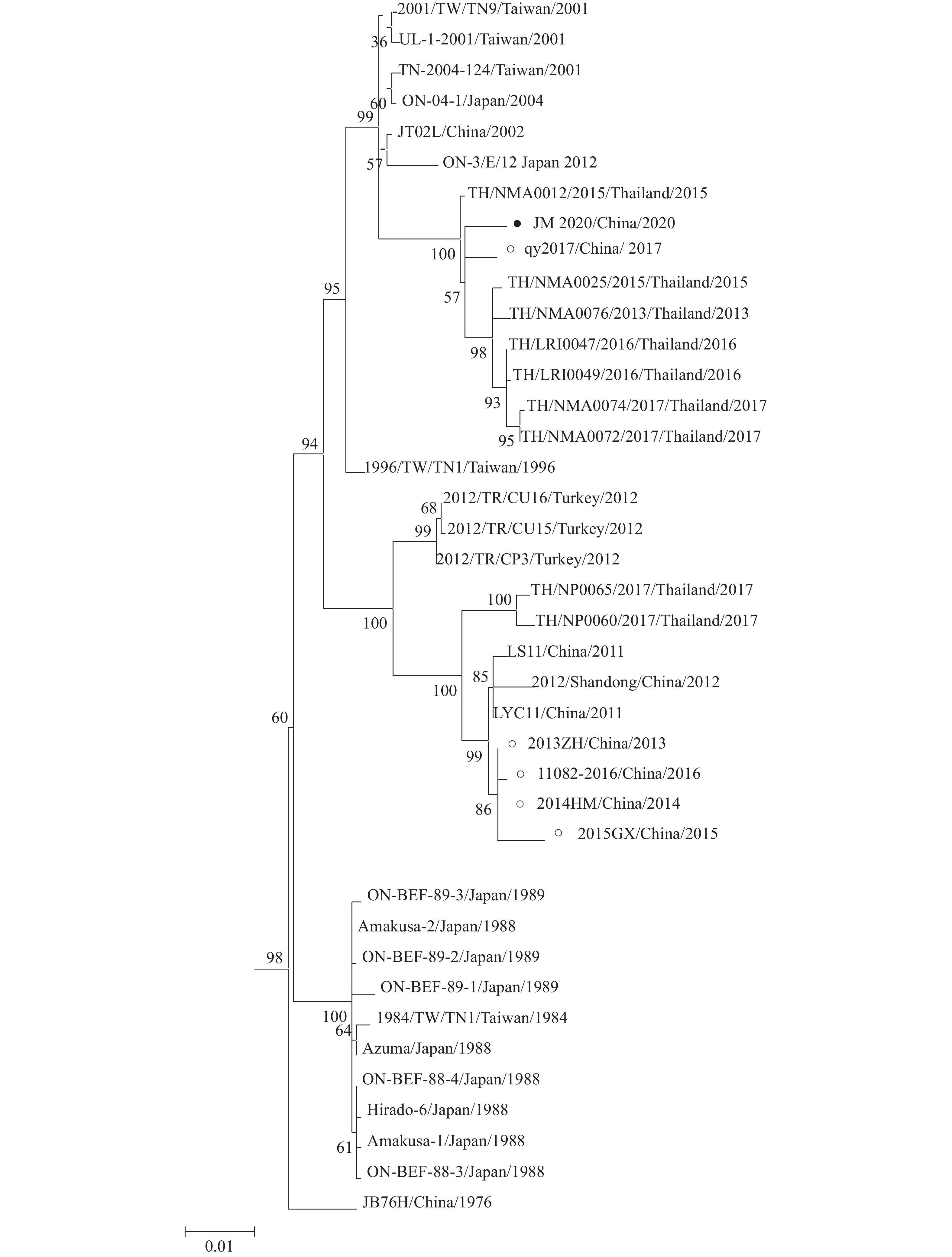
图 6 JM 2020毒株全基因组进化树
“●”为JM 2020毒株
Figure 6. Phylogenetic tree of whole genome of JM 2020 strain
“●” is the JM 2020 strain
![]()
图 7 JM 2020毒株P′基因序列分析
“.”为与对照组毒株氨基酸相同;“-”为该位置氨基酸缺失
Figure 7. Analysis of P′ gene sequence of JM 2020 strain
“.” is the same amino acid as the control group; “-” is the amino acid deletion at this position
表 1 扩增G基因以及JM 2020全基因组序列引物
Table 1 Primers employed for the amplication of G gene and whole genome of JM 2020 strain
扩增片段 Amplified fragment 名称 Name 序列(5′→3′) Sequence 位置/bp Location 产物大小/bp Size of product G BEFV G F ATGTTCAAGGTCCTCATAATTACC 3 059~3 082 1 872 BEFV G R TTAATGATCAAAGAACCTATCATCACCGATTGGTTTAC 4 893~4 930 1 BEFV1F ACGAGAAAAAACAAAAAAAC 1~20 820 BEFV1R GATACAAAAATCCAATCAAG 801~820 2 BEFV2F ACAAGGATGCCGCAGGAC 730~747 2 120 BEFV2R TTCCCTCTTCTTTTGCTC 2 832~2 849 3 BEFV3F ATTCATCCTAGAGATCAGG 2 737~2 752 1 455 BEFV3R ACCATTCCCCCTCTTGTTG 4 173~4 191 4 BEFV4F CTCTGTTTGTCCTTACCAG 4 112~4 127 2 066 BEFV4R AATGACAGTATCTTCATCAG 6 158~6 177 5 BEFV5F TAGCCAGGCAAGCATCCTG 6 002~6 019 2 196 BEFV5R TGGATTACTCTCTTGATCAAACATGC 8 170~8 197 6 BEFV6F AGTGAGATAGATATATGTGATATTCC 7 898~7 823 864 BEFV6R TGCAAATATATGATGAGAATCTTC 8 738~8 761 7 BEFV7F ATGAAGAGGAGTCCATGGAC 8 520~8 539 1 275 BEFV7R TGGGCCAAGTATTATGACTG 9 775~9 794 8 BEFV8F ATTTGGACATAAGTGGACGGTG 9 099~9 120 1 904 BEFV8R TCCATTAATTGCATCATGGGGTC 10 980~11 002 9 BEFV9F CAGACAATCAGCATGTG 10 643~10 659 2 181 BEFV9R TACAGTTCCTTGAAGTGC 12 806~12 823 10 BEFV10F AAACTTGACTCATCGCAG 12 758~12 775 2 185 BEFV10R ACGAAGAAAAACAAATAAAATAC 14 920~14 942  下载: 导出CSV
下载: 导出CSV
表 2 JM 2020单个基因编码区之间的起始信号和终止信号
Table 2 Initiation signal and termination signal between individual gene coding region of JM 2020 strain
基因 Gene 起始信号序列 Initiation signal sequence 终止信号序列 Termination signal sequence 编码氨基酸数/aa Number of coded amino acids ORF位置/bp ORF position 基因间隔核苷酸数/nt Nucleotide number of gene interval N AACAGG CATGAAAAAAA 431 72~1 367 21 P AACAGG CATGAAAAAAA 278 1 415~2 251 47 M AACAGG CATGAAAAAAA 223 2 320~2 991 68 G AACAGG CATGAAAAAAA 623 3 059~4 930 67 GNS AACAGG CATGAAAAAAA 603 4 954~6 765 23 α1、α2 AACAGG CTTGAAAAAAA 88、116 6 830~7 096、7 098~7 448 64 β AACAGG CATGAAAAAAA 147 7 508~7 951 59 γ AACAGG CATGAAAAAAA 114 8 031~8 375 79 L AACAGG CATGAAAAAAA 2 147 8 411~14 854 35
下载: 导出CSV
表 3 JM 2020与参考毒株各基因编码区氨基酸序列相似性
Table 3 Amino acid sequence similarity of individual gene coding region between JM 2020 and reference strains
% 参考毒株 Reference strain N P M GNS G L Bovine/China/Henan1/2012 99.5 97.8 100.0 76.4 98.6 98.2 qy2017/China/2017 99.5 99.3 88.0 99.5 99.0 99.7 Australian NC 002526.1 97.9 84.5 96.9 92.7 94.4 96.5 Australian AF234533.1 97.9 84.5 96.9 92.7 94.4 96.5 BEFV/Israel /2006 99.1 87.4 98.2 95.2 96.1 97.5 JT02L/China/2002 99.3 97.8 99.6 99.0 98.4 99.3 TH/LRI0045/East Asia/2016 99.5 98.6 100.0 99.7 99.2 99.7 India/2019 99.1 87.1 87.2 94.2 96.3 97.3 RSA/OBP/BEF2008/South Africa /2008 99.1 78.1 87.2 86.9 93.1 96.5 BEFV/Ad12/TUR/2012 98.6 87.1 98.2 93.2 96.5 97.3
下载: 导出CSV
-
[1] WALKER P J. Bovine ephemeral fever in Australia and the world[J]. Current Topics in Microbiology and Immunology, 2005, 292: 57-80.
[2] 刘和斯. 牛流行热的临床特点、症状及防治措施[J]. 畜禽业, 2021, 32(10): 139. [3] LEE F. Bovine ephemeral fever in Asia: Recent status and research gaps[J]. Viruses, 2019, 11(5): 412. doi: 10.3390/v11050412.
[4] JIA K, JIA R R, WANG X B, et al. Genetic characterization of bovine ephemeral fever virus in southern China, 2013−2017[J]. Virus Genes, 2020, 56(3): 390-395. doi: 10.1007/s11262-020-01740-w
[5] LIN G Z, QIU C Q. Phylogenetic relationships of the partial G gene sequence of bovine ephemeral fever virus isolated from Mainland China, Taiwan, Japan, Australia, Turkey and Israel[J]. Journal of Animal and Veterinary Advances, 2012, 11(17): 3217-3222. doi: 10.3923/javaa.2012.3217.3222
[6] STOKES J E, DARPEL K E, GUBBINS S, et al. Investigation of bovine ephemeral fever virus transmission by putative dipteran vectors under experimental conditions[J]. Parasites & Vectors, 2020, 13(1): 597. doi: 10.1186/s13071-020-04485-5.
[7] 李成, 谷守林, 姜绍德, 等. 应用电镜技术对牛流行热病毒形态学的研究[J]. 电子显微学报, 1993(1): 35. [8] 江船. 牛流行热病毒感染宿主细胞差异表达miRNA的筛选及其鉴定[D]. 济南: 山东大学, 2018. [9] LIU D, LI K, ZHANG L, et al. Seroprevalence investigation of bovine ephemeral fever in yaks in Tibetan Plateau of China from 2012 to 2015[J]. Tropical Animal Health and Production, 2017, 49(1): 227-230. doi: 10.1007/s11250-016-1172-9
[10] LI Z, ZHENG F, GAO S, et al. Large-scale serological survey of bovine ephemeral fever in China[J]. Veterinary Microbiology, 2015, 176(1/2): 155-160.
[11] 汪祥斌, 贾坤, 贾荣荣, 等. 牛流行热病毒TaqMan实时定量PCR检测方法的建立及应用[J]. 中国兽医学报, 2020, 40(3): 491-495. [12] WANG F I, HSU A M, HUANG K J. Bovine ephemeral fever in Taiwan[J]. Journal of Veterinary Diagnostic Investigation, 2001, 13(6): 462-467. doi: 10.1177/104063870101300602
[13] TRINIDAD L, BLASDELL K R, JOUBERT D A, et al. Evolution of bovine ephemeral fever virus in the Australian episystem[J]. Journal of Virology, 2014, 88(3): 1525-1535. doi: 10.1128/JVI.02797-13
[14] CHAISIRIRAT T, SANGTHONG P, ARUNVIPAS P, et al. Molecular characterization of bovine ephemeral fever virus in Thailand between 2013 and 2017[J]. Veterinary Microbiology, 2018, 227: 1-7. doi: 10.1016/j.vetmic.2018.10.013
[15] 童树喜. 牛流行热的流行病学调查及防治[J]. 中国畜牧业, 2021(5): 72-73. doi: 10.3969/j.issn.2095-2473.2021.05.035 [16] WALKER P J, KLEMENT E. Epidemiology and control of bovine ephemeral fever[J]. Veterinary Research, 2015, 46: 124. doi: 10.1186/s13567-015-0262-4.
[17] ZHENG F, QIU C. Phylogenetic relationships of the glycoprotein gene of bovine ephemeral fever virus isolated from mainland China, Taiwan, Japan, Turkey, Israel and Australia[J]. Virology Journal, 2012, 9: 268. doi: 10.1186/1743-422X-9-268.
[18] AZIZ-BOARON O, KLAUSNER Z, HASOKSUZ M, et al. Circulation of bovine ephemeral fever in the Middle East: Strong evidence for transmission by winds and animal transport[J]. Veterinary Microbiology, 2012, 158(3/4): 300-307.
[19] 杨金雨, 李赞, 王丹. 重新认识牛流行热及其疫苗[J]. 中国奶牛, 2018(5): 47-49.