
Cloning, subcellular localization and functional analysis of GmMADS4 in soybean
-
摘要:目的
挖掘大豆Glycine max MADS转录因子家族成员GmMADS4基因信息,分析其结构及功能。
方法通过生物信息学分析,对GmMADS4基因进行基因结构、编码蛋白信息、保守结构域、系统进化树以及互作蛋白预测等分析。利用烟草叶片瞬时转化法分析亚细胞定位,通过RT-qPCR进行组织部位及响应缺素的表达模式分析,利用下胚轴复合植株转化法分析超量表达GmMADS4对转基因毛根生长的影响。
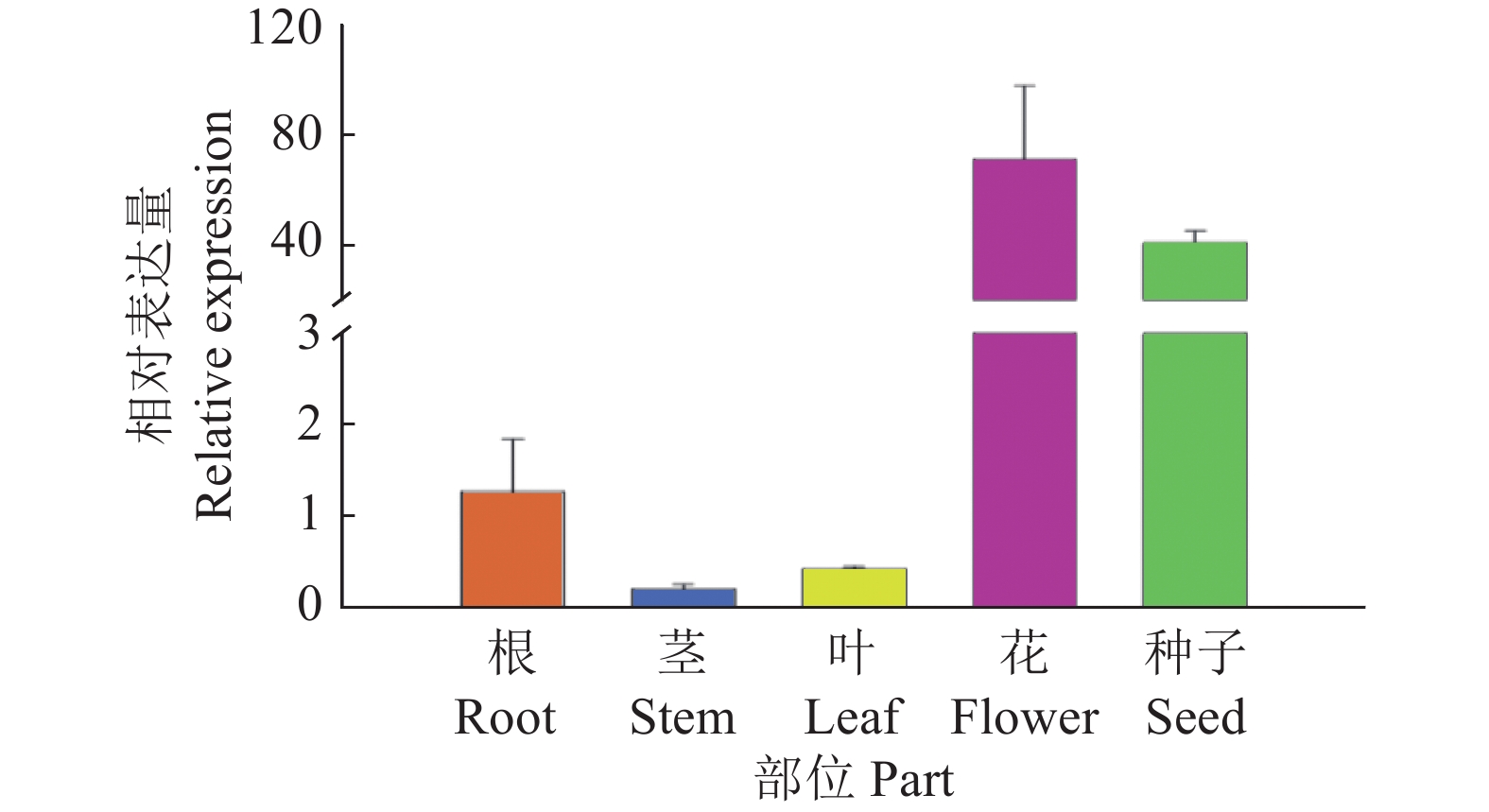
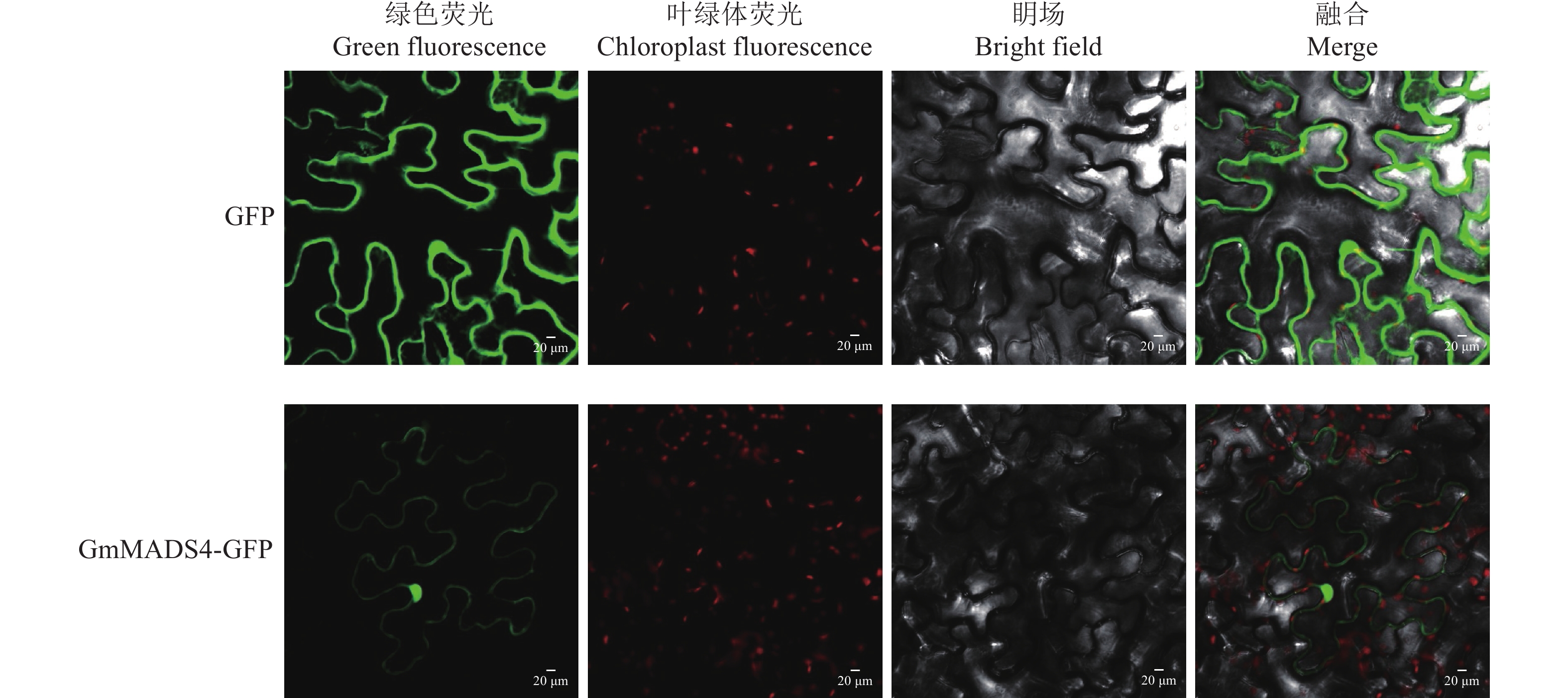
结果GmMADS4基因开放阅读框长732 bp,编码蛋白相对分子质量为28 000;保守结构域含有MADS-box和K-box,属于II型MADS家族成员,与拟南芥的AtAP3相似性较高;GmMADS4在大豆多个部位均有表达,且在花和种子中的表达量较高;缺氮和缺磷处理均显著增加GmMADS4在叶和根部的表达量;GmMADS4主要定位在细胞核,超量表达GmMADS4显著增加转基因毛根的可溶性磷含量。
结论GmMADS4属于大豆II型MADS家族成员,具有核定位功能,可能在大豆种子和花的发育过程中发挥作用,并参与大豆根部缺磷响应及磷稳态调节。
Abstract:ObjectiveTo explore the genetic information of GmMADS4, a member of soybean (Glycine max) MADS transcription factor family, and analyze its structure and function.
MethodThe structure, coding protein information, conserved domain, phylogenetic tree analysis and interaction protein prediction of GmMADS4 were analyzed by bioinformatics analysis. Transient transformation of tobacco leaves was used to analyze subcellular localization, RT-qPCR was used to analyze its expression patterns in different tissues and response to nutrient deficiency, and hypocotyl complex plant transformation method was used to analyze the effects of overexpression of GmMADS4 on transgenic hair root growth.
ResultThe length of open reading frame of GmMADS4 was 732 bp and the relative molecular mass of coding protein was 28 000. The conserved domain of GmMADS4 contained MADS-box and K-box, which was a member of type II MADS family, and had a high similarity with Arabidopsis AtAP3. GmMADS4 was expressed in many parts of soybean, especially in flowers and seeds. The expression of GmMADS4 in leaves and roots significantly increased by nitrogen and phosphorus deficiency induction. GmMADS4 was mainly located in the nucleus, and overexpression of GmMADS4 significantly increased soluble phosphate content in transgenic hair roots.
ConclusionGmMADS4 is a member of soybean type II MADS family, and has nuclear localization function, which may play a role in the development of soybean seeds and flowers, as well as participate in soybean root adaptive responses to phosphorus deficiency and the regulation of phosphate homeostasis.
-
Keywords:
- Soybean /
- GmMADS4 /
- Subcellular localization /
- Expression pattern /
- Functional analysis
-
染色体作为生物遗传物质的载体,对当前生命遗传研究开展尤为重要。染色体消除技术(Chromosome elimination technology)近年来随着基因编辑技术的发展不断进步,在动物模型构建、家畜育种改良中的性别控制、特定染色体的功能研究以及人类非整倍体疾病治疗等领域具有重要的应用价值。目前通过该技术已成功在人、小鼠、斑马鱼等物种上实现了特定染色体的消除[1-3],然而打靶效率低、非靶点的额外失活及操作过程的繁琐性大大制约了该技术的发展[3-5]。但随着新一代CRISPR/Cas9技术的出现,灵活的靶点选择、较高的编辑效率以及对于靶点多重切割的可能性使该技术的有效运用成为可能。
目前,研究者们已通过锌指核酸酶(Zinc finger nuclease,ZFN)技术、Cre/loxP系统、TKNEO基因敲入等多种方法实现了目标染色体的敲除[5-7]。ZFN技术通过人工改造的核酸内切酶(锌指蛋白单元)与特定的DNA序列结合赋予其精确的打靶功能[8],但其脱靶效应、毒性及锌指结构的低适用性造成其基因操作效率低下[9];Cre/loxP系统主要由Cre重组酶与2个loxP位点组成,由于loxP的位点不同会产生DNA链的插入、删除、易位等不同重组方式[10],但该系统的使用需要引入特定表达元件且本身会对细胞产生毒性[4, 11];TKNEO是胸苷激酶(Thymidine kinase,TK)和新霉素抗性(Neomycin resistance,NEO)的融合编码基因,利用传统基因打靶技术敲入目标染色体进行药筛可产生自发性染色体缺失[3],但其操作的繁琐性和不确定性使其并不具有广泛的适用性。
CRISPR/Cas9技术近年来已经成为了基因编辑的主要手段,其由大肠埃希菌Ⅱ型CRISPR/Cas系统改造而来,由于其引导RNA(Small guide RNA,sgRNA)序列的灵活性而能对多个位点进行灵活编辑,从而实现基因的定点敲入、敲除或其他多种修饰[12-16]。该系统主要由2个部分组成,Cas9蛋白和sgRNA,其中后者负责识别目标靶序列,前者依靠其自身含有的2个切割域完成对目标序列的切割,之后依靠细胞本身的2种修复机制(同源重组修复和非同源末端连接)实现目标序列的精确编辑[17-21]。最新研究表明,CRISPR/Cas9系统可通过产生多重DNA双链断裂(Double-strand breakage,DSB)引起Y染色体降解[22-23],对这种方法,虽然部分细胞会因自身修复机制保持基因组的稳定性,但是仍有相当的几率可产生特定染色体消除的细胞,通过筛选出此种染色体消除的细胞系作为供体应用于动物克隆,可制备出特定染色体消除的动物,因此达到控制动物本身及其后代的性别进行偏移的目的。
本研究拟通过CRISPR/Cas9技术,在猪Y染色体内寻找能够被特异sgRNA识别的重复序列,进行多位点切割以实现目标染色体的降解。本研究旨在为特定染色体及其携带基因功能的研究提供一种新的工具,同时也为实现大型家畜性别控制提供一种新的技术方案。
1. 材料与方法
1.1 材料
本试验所用的CRISPR/Cas9载体HP180_px330_GFP及猪胎儿成纤维细胞(Porcine embryonic fibroblast,PEF)由国家生猪种业工程技术研究中心保存。设计的sgRNA及相关引物由华大基因合成。质粒与基因组DNA抽提试剂盒、限制性内切酶BpiI、gRNA体外转录试剂盒GeneArtTM Precision gRNA Synthesis Kit、荧光定量试剂盒PowerUpTMSYBRTMGreen Master Mix购自赛默飞公司;T4 DNA连接酶购自TaKaRa公司;Cas9内切酶购自NEB公司;感受态细胞Trans5α、6×DNA Loading buffer、RNA loading buffer购自全式金生物。高糖DMEM 培养液、胰酶、胎牛血清均购自Gibco公司。
1.2 方法
1.2.1 寻找Y染色体特异靶向重复序列及sgRNA设计
在Ensembl获取整个Y染色体注释文件,在Linux下用awk命令提取注释文件中的所有基因,并在Ensembl上将每个基因与全基因组序列进行逐一比对,最终筛选出符合条件的多拷贝基因。
利用在线网站CCTop - CRISPR/Cas9 target online predictor( https://cctop.cos.uni-heidelberg.de/)对所选择靶基因进行sgRNA设计,确定最终序列与位点,并送至华大基因合成。
1.2.2 sgRNA体外切割验证
对设计好的sgRNA体外转录进行切割,初步筛选出效率较高的进行载体构建。首先进行靶片段的PCR扩增。根据本试验靶基因的选择,使用Primer Premier 6进行相应靶点引物设计,合成目标靶片段。然后根据赛默飞公司的GeneArtTM Precision gRNA Synthesis Kit说明书设计sgRNA体外转录模板引物,并最终进行Gene_ID为ENSSSCG00000043058、ENSSSCG00000046447的转录模板合成(以下简称为43058、46447)。其合成原理(图1)及相关引物(表1、表2)如下所示。
![]() 图 1 sgRNA合成原理图体外合成的gRNA由2条34~38 bp寡核苷酸与tracrRNA片段退火形成,其中Target-F由T7启动子和靶序列的16~20 bp组成,Target-R由tracrRNA片段的部分序列和与目标序列互补的19~20 bp组成Figure 1. Schematic diagram of sgRNA synthesisTwo 34−38 bp oligonucleotides are needed to assemble the synthetic gRNA with tracrRNA fragment, among them the Target-F is composed of the T7 promoter and 16−20 bp sequence of the target sequence, and the Target-R is composed of partial sequence of tracrRNA fragment and 19−20 bp sequence complementary to the target sequence表 1 靶片段扩增引物Table 1. Primers of target fragment amplification
图 1 sgRNA合成原理图体外合成的gRNA由2条34~38 bp寡核苷酸与tracrRNA片段退火形成,其中Target-F由T7启动子和靶序列的16~20 bp组成,Target-R由tracrRNA片段的部分序列和与目标序列互补的19~20 bp组成Figure 1. Schematic diagram of sgRNA synthesisTwo 34−38 bp oligonucleotides are needed to assemble the synthetic gRNA with tracrRNA fragment, among them the Target-F is composed of the T7 promoter and 16−20 bp sequence of the target sequence, and the Target-R is composed of partial sequence of tracrRNA fragment and 19−20 bp sequence complementary to the target sequence表 1 靶片段扩增引物Table 1. Primers of target fragment amplification引物名称 Primer name 引物序列(5′→3′) Primer sequence 产物长度/bp Product length 43058target_F GTCCCATTTTCCAGGCCTTA 616 43058target_R GTTCAGAGGCACACAATGTG 46447target_F AGTTGAAGGCCACTTGGTCA 657 46447target_R ATGACAATTTGTCCTGGTGGCC 表 2 sgRNA体外转录模板合成引物Table 2. Primers for synthesis of sgRNA in vitro transcription template引物名称 Primer name 引物序列(5'→3') Primer sequence T43058-F1 TAATACGACTCACTATAGTCACTGGGGACTTGGACTT T43058-R1 TTCTAGCTCTAAAACCAAGTCCAAGTCCCCAGTG T43058-F2 TAATACGACTCACTATAGGTGCATCTTTAGGAGACAC T43058-R2 TTCTAGCTCTAAAACGTGTCTCCTAAAGATGCACC T46447-F1 TAATACGACTCACTATAGATGTGGCTGCAGAAACTCG T46447-R1 TTCTAGCTCTAAAACCGAGTTTCTGCAGCCACAT T46447-F2 TAATACGACTCACTATAGAAGAAGCGCTCTAGAACAGG T46447-R2 TTCTAGCTCTAAAACCCTGTTCTAGAGCGCTTCT 体外Cas9切割验证主要包含3个过程:Cas9-sgRNA的自发组装、Cas9-sgRNA复合体与靶片段的特异结合、Cas9-sgRNA对靶点进行特异切割。采用20 μL反应体系:体外转录sgRNA模板100 ng,靶片段250 ng,Cas9蛋白1 μL,10×Cas9 Reaction buffer 2 μL,最后加Nuclease-free water补足至20 μL。
在进行Cas9体外切割时,反应体系首先加入除靶片段外其他成分进行Cas9与sgRNA组装(组装时间30 min),再加入相应靶片段孵育1 h,切割完毕后在反应体系中加入2 μL的蛋白酶K再次孵育20 min。最终切割产物加入6×DNA Loading Buffer进行20 g/L的凝胶电泳,分析电泳条带灰度计算切割效率。
1.2.3 CRISPR/Cas9载体构建
确定有效的sgRNA后进行真核细胞表达载体构建。首先使用限制性内切酶BpiI对载体HP180_px330_GFP(图2)双酶切使其线性化,合成的sgRNA经退火形成双链由T4连接酶在16 ℃条件下连接1 h,然后热应激转入Trans5α感受态细胞,于220 r/min、37 ℃培养1 h后涂布含氨苄青霉素的LB平板,隔夜培养后挑取单克隆,进行菌液PCR并送测序,测序正确菌液进行质粒抽提,用于PEF电转。
1.2.4 PEF培养与转染
试验所用细胞经鉴定为公猪细胞(使用引物F:TCATAGCTCAAACGATGGACGTG,R:ACACAATGAAAGCGTTCATGGGTC,扩增片段大小为92 bp)。PEF复苏后加入含体积分数为12%胎牛血清的高糖DMEM培养液,置于38 ℃、CO2体积分数为5%的培养箱中培养。当复苏后的细胞融合度达到80%~90%时,用质量分数为0.05%的胰酶消化后进行细胞计数。取1×106 mL−1细胞悬液加入1.5 mL离心管,加入100 μL电转液及目的质粒以Lonza核转仪进行电转,电转程序A003。转染24 h后,荧光显微镜下拍照并观察转染细胞荧光信号强弱,判断转染效果。
1.2.5 流式分选及单细胞克隆团鉴定
细胞转染2~3 d后进行流式分选(Fluorescence activating cell sorter,FACS)。细胞分选前经质量分数为0.05%的胰酶消化,加入含体积分数为2%胎牛血清的 DPBS重悬,并在上机前经40 µm细胞筛过滤防止细胞团块的干扰。根据GFP标记激发波长488 nm分选出阳性细胞(5×104~10×104个)并进行极限稀释(梯度稀释降低细胞密度)培养,细胞培养形成单个细胞团后取部分细胞抽提DNA进行Y染色体性别决定区(Sex determination region of Y chromosome, SRY)基因鉴定。引物信息如表3所示。
表 3 靶基因及SRY基因PCR引物Table 3. PCR primers of target gene and SRY gene引物名称 Primer name 引物序列(5′→3′) Primer sequence 产物长度/bp Product length 43058_F CATTGTGATGCATGGGCGATTT 238 43058_R GAGACCTCAAGTCCAAGTCCC 46447_F ATGTGGCTGCAGAAACTCGT 169 46447_R ATTTGTCCTGGTGGCCTGAC SRY_F GTGGCTGGGATGCAAGTGGAAA 228 SRY_R GCCTTGGCGACTGTGTATGTGAA 1.2.6 实时定量荧光PCR检测Y染色体敲除效率
分选后待细胞融合度达到80%~90%提取基因组DNA,并设计相应靶基因及SRY定量引物(表3)观察其DNA含量是否显著降低。以基因组为模版检测目的基因在DNA水平的含量是否变化,并以基因组水平的β-actin为内参。反应体系与反应产物均按照相应试剂盒(PrimeScriptTM RT reagent Kit with gDNA Eraser与PowerUpTM SYBRTM Green Master Mix)说明进行。
1.2.7 核型鉴定
对SRY鉴定为阴性细胞继续扩繁培养,并传代至T25培养瓶,待细胞生长至融合度为70%~80%时进行核型分析,判断染色体核型是否缺失。核型分析由杭州卡优太谱生物科技公司进行。
2. 结果与分析
2.1 多拷贝基因查询及sgRNA设计结果
2.1.1 多拷贝基因查询结果
通过Ensembl的blast筛选出了35个位于Y染色体上的多拷贝基因,统计结果如表4所示。
表 4 猪Y染色体多拷贝基因信息1)Table 4. Muti-copy genes in Y chromosome of pig基因编号 Gene_ID 起始位置 Begin position 结束位置 End position 长度/bp Length 拷贝数 Copy number ENSSSCG00000041799 20570323 20570724 402 90 ENSSSCG00000045041 23748599 23749006 408 42 ENSSSCG00000043614 22488485 22488925 441 39 ENSSSCG00000043058 21558744 21559151 408 39 ENSSSCG00000050250 20115553 20117373 1821 32 ENSSSCG00000049276 20156946 20158766 1821 32 ENSSSCG00000041211 20259563 20261383 1821 32 ENSSSCG00000041323 20851299 20853119 1821 32 ENSSSCG00000051247 23675355 23677155 1821 32 ENSSSCG00000044850 22938437 22940148 1721 32 ENSSSCG00000035359 19637403 19639087 1685 32 ENSSSCG00000038218 20710734 20712418 1685 32 ENSSSCG00000031994 20902449 20904133 1685 32 ENSSSCG00000032210 21694455 21696139 1685 32 ENSSSCG00000033039 19929688 19931370 1683 32 ENSSSCG00000044818 21140962 21142262 1301 32 ENSSSCG00000039954 22859918 22861602 1685 31 ENSSSCG00000049743 23367269 23368953 1685 31 ENSSSCG00000042334 19885774 19887455 1682 31 ENSSSCG00000042971 21982938 21984619 1682 31 ENSSSCG00000045651 23897691 23899366 1676 31 ENSSSCG00000051215 20442628 20444448 1821 30 ENSSSCG00000041507 22640161 22641981 1821 30 ENSSSCG00000045761 24007552 24009372 1821 29 ENSSSCG00000041750 23944189 23946009 1821 29 ENSSSCG00000050201 21301627 21303492 1821 29 ENSSSCG00000048908 19541113 19542933 1821 27 ENSSSCG00000045241 21443949 21445655 1707 26 ENSSSCG00000046033 23455676 23457401 1726 24 ENSSSCG00000036112 22537239 22541180 3942 3 ENSSSCG00000043960 23320436 23322365 1930 2 ENSSSCG00000031258 21822265 21828538 6274 * ENSSSCG00000051413 21205077 21216797 11721 * ENSSSCG00000046447 22138546 22177691 39146 * ENSSSCG00000042484 23810416 23850459 40244 * 1) “*”代表基因序列太长,不存在完全拷贝片段,拷贝数统计不完整,但存在部分序列多拷贝,因此也可作为多拷贝基因的一种;加粗Gene_ID为靶基因 1) “*” represents that the gene sequence is too long, there is no complete copy fragment, and the copy number statistics are incomplete, but there are multiple copies of some sequences, so it can also be used as a kind of multi copy gene;Bold gene ID represents the target gene 通过再次与NCBI数据库进行比对,以及基于靶点基因组测序的真实结果与sgRNA设计的实际情况,我们最终选择了网站预测评分较高、符合测序结果且在Y染色体上重复拷贝数靠前(多位点DSB)的Gene_ID为ENSSSCG00000043058、ENSSSCG00000046447的基因进行了sgRNA设计。这2个基因在全基因组的比对结果如图3所示。
![]() 图 3 ENSSSCG00000043058和ENSSSCG00000046447在猪染色体上的位置示意图所选择的靶基因仅存在于Y染色体上并有多个拷贝,有利于实现Y的多位点切割Figure 3. Locations of ENSSSCG00000043058 and ENSSSCG00000046447 on pig chromosomeThe selected target genes only exist on the Y chromosome and have multiple copies, which is conducive to the realization of Y multipoint cutting
图 3 ENSSSCG00000043058和ENSSSCG00000046447在猪染色体上的位置示意图所选择的靶基因仅存在于Y染色体上并有多个拷贝,有利于实现Y的多位点切割Figure 3. Locations of ENSSSCG00000043058 and ENSSSCG00000046447 on pig chromosomeThe selected target genes only exist on the Y chromosome and have multiple copies, which is conducive to the realization of Y multipoint cutting2.1.2 sgRNA设计结果
对ENSSSCG00000043058和ENSSSCG00000046447位点进行sgRNA设计(表5)。在sgRNA及其互补序列末端分别加上与BpiI酶切HP180_px330_GFP质粒互补的黏性末端,由华大基因合成相应的Oligo。
表 5 sgRNA靶点位置和序列Table 5. Location and sequence of sgRNA targetsgRNA Y染色体位置 Position on Y chromosome 靶点序列 Target sequence 43058sg1 21558841~21558860 TCACTGGGGACTTGGACTTG 43058sg2 21558795~21558814 ATGGAGGATTCTGAACCAGT 46447sg1 22254580~22254599 GATGTGGCTGCAGAAACTCG 46447sg2 22254625~22254644 AAGAAGCGCTCTAGAACAGG 2.2 sgRNA体外切割结果
以野生型PEF DNA为模板进行PCR扩增,产物进行凝胶电泳分析后切胶回收得到sgRNA体外转录模板。根据GeneArtTM Precision gRNA Synthesis Kit试剂盒的体系与方法进行sgRNA的转录模板合成(图4A)与体外sgRNA转录(图4B),转录产物经纯化后电泳验证条带完整性(长度100 bp),电泳结果与预期一致。
![]() 图 4 靶片段模板DNA扩增与sgRNA体外转录结果M:DL2000 DNA marker;1和2分别代表扩增靶片段43058和46447,长度分别为616和657 bp;3、4、5和6代表转录产物43058sg1、43058sg2、46447sg1和46447sg2,长度均为100 bpFigure 4. Amplification of target fragment template DNA and in vitro transcription of sgRNAM:DL2000 DNA marker; 1 and 2 represent the amplified target fragment 43058 and 46447 respectively, and the lengths are 616 and 657 bp respectively; 3, 4, 5 and 6 represent transcripts 43058sg1, 43058sg2, 46447sg1 and 46447sg2 respectively, and the lengths all are 100 bp
图 4 靶片段模板DNA扩增与sgRNA体外转录结果M:DL2000 DNA marker;1和2分别代表扩增靶片段43058和46447,长度分别为616和657 bp;3、4、5和6代表转录产物43058sg1、43058sg2、46447sg1和46447sg2,长度均为100 bpFigure 4. Amplification of target fragment template DNA and in vitro transcription of sgRNAM:DL2000 DNA marker; 1 and 2 represent the amplified target fragment 43058 and 46447 respectively, and the lengths are 616 and 657 bp respectively; 3, 4, 5 and 6 represent transcripts 43058sg1, 43058sg2, 46447sg1 and 46447sg2 respectively, and the lengths all are 100 bp将扩增好的靶片段、sgRNA按照一定比例与Cas9混合,按照方法中的步骤进行反应得到切割产物,43058sg1的理论切割产物长度为525和91 bp,43058sg2的理论切割产物长度为444和172 bp,46447sg1的理论切割产物长度为498和159 bp,46447sg2的理论切割产物长度为453和204 bp。凝胶电泳分析表明43058sg2与46447sg2均达到了预期的切割效果,并且43058sg2实现了接近完全的靶基因切割(图5)。灰度分析表明,46447sg2的体外切割效率也达到了58%(图6),最终我们选择 43058sg2和46447sg2进行载体构建。
![]() 图 5 设计的sgRNA体外切割电泳图M:DL7000 DNA marker;1、4:未转染细胞对照;2、3、5、6为设计的43058sg1、43058sg2、46447sg1以及46447sg2的实际切割产物Figure 5. Electrophoregram of in vitro cleavage of designed sgRNAM: DL7000 DNA marker; 1, 4: Control of untransfected cells; 2, 3, 5, 6 are the actual cutting products of designed 43058sg1, 43058sg2, 46447sg1 and 46447sg2 respectively
图 5 设计的sgRNA体外切割电泳图M:DL7000 DNA marker;1、4:未转染细胞对照;2、3、5、6为设计的43058sg1、43058sg2、46447sg1以及46447sg2的实际切割产物Figure 5. Electrophoregram of in vitro cleavage of designed sgRNAM: DL7000 DNA marker; 1, 4: Control of untransfected cells; 2, 3, 5, 6 are the actual cutting products of designed 43058sg1, 43058sg2, 46447sg1 and 46447sg2 respectively2.3 HP180_px330_GFP载体构建结果
HP180_px330_GFP骨架上有预留的BpiI双酶切位点,其线性化后与退火形成具有黏性末端的双链sgRNA,在连接酶作用下形成闭合环状结构。其测序结果如图7所示,结果与设计的sgRNA结果一致,表明载体构建成功。
![]() 图 7 HP180_px330_GFP质粒测序结果绿色下划线部分为BpiI双酶切位点,红色箭头标注部分为针对各靶位点构建的识别序列Figure 7. HP180_ px330_ GFP plasmid sequencing resultsThe underlined part in green is BpiI double digestion site, and the marked part in red arrow is the recognition sequence constructed for each target site
图 7 HP180_px330_GFP质粒测序结果绿色下划线部分为BpiI双酶切位点,红色箭头标注部分为针对各靶位点构建的识别序列Figure 7. HP180_ px330_ GFP plasmid sequencing resultsThe underlined part in green is BpiI double digestion site, and the marked part in red arrow is the recognition sequence constructed for each target site2.4 细胞性别鉴定及细胞转染检测
2.4.1 性别鉴定
对试验所用PEF进行SRY鉴定,电泳结果显示目的细胞为雄性(图8)。
![]() 图 8 SRY基因检测结果M:DL700 DNA marker;1:空白水对照;2:阴性对照母猪DNA;3:试验所用公猪细胞Figure 8. SRY gene assay resultM:DL700 DNA marker;1: Blank water control; 2: DNA of negative control sow; 3: Boar cells used in the experiment
图 8 SRY基因检测结果M:DL700 DNA marker;1:空白水对照;2:阴性对照母猪DNA;3:试验所用公猪细胞Figure 8. SRY gene assay resultM:DL700 DNA marker;1: Blank water control; 2: DNA of negative control sow; 3: Boar cells used in the experiment2.4.2 细胞转染
将构建的CRISPR/Cas9载体通过细胞核转染导入雄性PEF中。由于载体带有增强绿色荧光蛋白(Enhanced green fluorescent protein,EGFP)表达单元,可通过观察EGFP的表达评估载体的转染效率。对已电转的细胞培养24 h后观察荧光信号,确定已构建好的载体成功电转进入细胞。结果(图9)表明质粒已成功表达。
![]() 图 9 PEF电转后荧光表达效果图A、B分别为2次重复试验的转染效果示意图;A1、B1为细胞明场图片,A2、B2分别为A1、B1同视野的EGFP表达图Figure 9. EGFP expression after PEF electroporationA and B are cell transfection results of two repeated experiments;A1 and B1 are transfected cells imaged in bright field, and A2 and B2 are EGFP expression images of the same vision as A1 and B1, respectively
图 9 PEF电转后荧光表达效果图A、B分别为2次重复试验的转染效果示意图;A1、B1为细胞明场图片,A2、B2分别为A1、B1同视野的EGFP表达图Figure 9. EGFP expression after PEF electroporationA and B are cell transfection results of two repeated experiments;A1 and B1 are transfected cells imaged in bright field, and A2 and B2 are EGFP expression images of the same vision as A1 and B1, respectively2.5 FACS分选阳性细胞及单克隆鉴定结果
通过FACS分选EGFP阳性细胞,我们发现对照组与试验组GFP信号存在显著差异,其中对照组未检测到GFP信号,而阳性对照的细胞转染效率达到了71%,这与荧光观察结果类似。通过培养EGFP阳性细胞,获得单细胞克隆团。接着我们对单克隆细胞团提取微量DNA,进行相应的SRY检测,发现1/3的细胞克隆未检测到SRY信号(图10),显示这些细胞的Y染色体存在缺失的可能。
![]() 图 10 单克隆细胞团SRY基因鉴定示意图M:DL700 DNA marker;1~24代表单克隆细胞团编号Figure 10. SRY gene identification of monoclonal cell clustersM: DL700 DNA marker; 1−24 represent the number of monoclonal cell clusters
图 10 单克隆细胞团SRY基因鉴定示意图M:DL700 DNA marker;1~24代表单克隆细胞团编号Figure 10. SRY gene identification of monoclonal cell clustersM: DL700 DNA marker; 1−24 represent the number of monoclonal cell clusters2.6 实时定量荧光PCR检测目的基因及SRY基因
由于选用的目的基因在PEF上并不表达mRNA,因此采用qPCR检测目的基因(内参基因选择β-actin)和SRY基因的DNA含量。我们抽提EGFP阳性细胞的基因组DNA,进行qPCR分析目的基因(43058和46477)和SRY在DNA水平的含量。qPCR结果显示在进行靶基因敲除后,靶基因含量均显著降低。其中靶基因43058和46447分别下降了约20%和30%(P<0.05),对应的SRY也下降了7%和11%。46447敲除效果相对于43058的基因敲除效果更为显著,不仅靶基因敲除效率较高,SRY基因DNA水平也下降较多(图11)。
![]() 图 11 EGFP阳性细胞中靶基因和SRY基因相较于正常雄性细胞的DNA水平检测“*”“**”“***”分别表示差异达到0.05、0.01和0.001的显著水平(t检验)Figure 11. Detection of DNA levels of target genes and SRY gene in EGFP-positive cells compared with normal male cells“*” “**” “***” indicate significant differences at 0.05, 0.01 and 0.001 levels respectively (t test)
图 11 EGFP阳性细胞中靶基因和SRY基因相较于正常雄性细胞的DNA水平检测“*”“**”“***”分别表示差异达到0.05、0.01和0.001的显著水平(t检验)Figure 11. Detection of DNA levels of target genes and SRY gene in EGFP-positive cells compared with normal male cells“*” “**” “***” indicate significant differences at 0.05, 0.01 and 0.001 levels respectively (t test)2.7 核型分析结果
核型鉴定结果显示上述流式分选得到的EGFP阳性细胞存在染色体缺失现象,表明设计的sgRNA在细胞水平上能够实现整个Y染色体的删除。在鉴定的12个单细胞核型中,有1个完全缺失Y染色体(图12)。
![]() 图 12 EGFP阳性细胞核型分析结果示意图Figure 12. Diagram of karyotype assay result of EGFP-positive cells
图 12 EGFP阳性细胞核型分析结果示意图Figure 12. Diagram of karyotype assay result of EGFP-positive cells3. 讨论与结论
CRISPR/Cas9近年来由于其基因编辑的灵活性和高效率而被广泛应用。其通过DSB导致的非保守非同源末端连接修复途径造成目的基因的敲除效果,但是该种途径常常造成异常的插入缺失,甚至破坏染色体和基因组的完整性[24-26]。而以天然的(或定向的)DNA模板的同源重组修复(Homologous recombination repair,HDR)途径则可以对目的基因位点进行精确修饰而产生意向的目的基因型[14, 27],本研究利用CRISPR系统的高效切割能力对Y染色体上多个重复基因位点进行切割,通过产生多重DSB损伤基因组和DNA修复的稳定性,引起Y染色体降解。实现整条染色体删除是建立在CRISPR/Cas9系统能对多个位点进行同时编辑的基础上的,虽然早期的研究已经证明能够产生大片段的缺失,但这对于整条染色体的消融还远远不够。2015年,Yang等[28]首先通过靶向内源性逆转率病毒(PERVs)的62个重复位点在猪上实现了PERVs的全基因组降解;随后,研究人员进一步利用CRISPR/Cas9对靶点切割的多重性,成功在ES细胞、体内细胞和受精卵中删除了Y染色体,并对其他常染色体(7号、14号)及X染色体进行了特异性消除[22-23]。实现该技术的关键是在Y染色体上寻找能够被sgRNA特异识别的靶向序列,这些序列在其他常染色体或X染色体上不能被识别,通过对其进行多重切割从而实现整条染色体的消融。因此在选择靶点时,单一数据库的比对结果不能很好地反映个体染色体组的真实情况,最终选择靶基因是在Ensembl比对的基础上重新与NCBI、Sanger测序结果进行比对,选择多个数据库共有序列并与个体基因组序列结合,增加靶点的可靠性。
不同于一般的sgRNA效率检测采用T7E1酶切和测序验证,由于本试验靶点为Y上的多拷贝重复序列,这些片段在Y上广泛分布且保守性不强,可能会存在一些SNP或者突变,导致扩增未编辑前的靶基因产物时出现套峰现象,和编辑后的测序结果无法区分。因此在这里只能通过体外切割进行sgRNA的效率检测。最终以ID为ENSSSCG00000043058、ENSSSCG00000046447的基因为靶点设计sgRNA,通过对Y上不同靶点,即散在重复和簇状聚集的靶基因的打靶,探索了不同打靶位点对编辑效率的影响,结果表明簇状聚集的打靶效果要明显优于散在重复序列,这与Zuo等[22]和Adikusuma等[23]的研究结果一致,其原因可能是长臂偶然断裂造成的染色体截断或易位导致的。另外由于我们选择的基因为非蛋白编码基因,所以在定量分析时最终选择以DNA为模板。在检测靶点DNA含量变化的同时我们也对SRY含量进行了定量,结果均表明其表达量随靶基因降低而降低,虽然这种变化并不明显,但从侧面说明了整条Y染色体降解的可能性。核型分析的结果也表明部分单细胞克隆在染色体水平确实存在着Y染色体的丢失,表明通过多位点断裂造成整条染色体的缺失是可行的。
本研究通过寻找能够特异性识别Y染色体的多拷贝重复序列设计sgRNA,并通过体外切割、qPCR、细胞转染、核型分析等手段证明了该系统在体外体内都有较高的切割效率,可成功实现整条染色体的消除,为未来染色体功能及动物性别控制研究提供了新的方向。
-
![]()
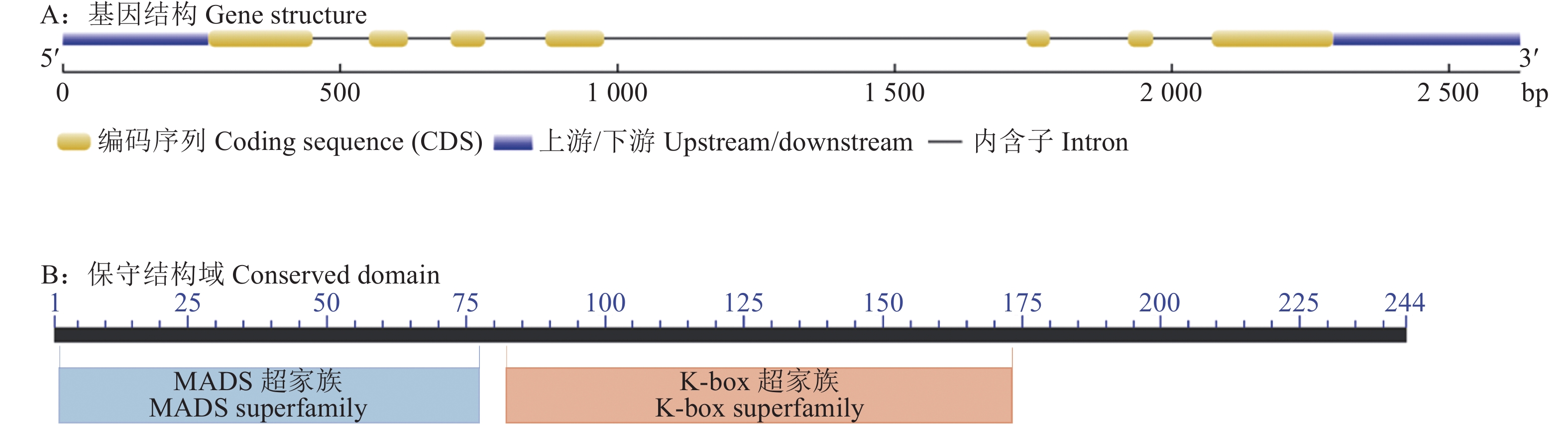
图 1 GmMADS4基因结构及保守结构域预测
Figure 1. Gene structure and conserved domain prediction of GmMADS4
![]()
图 2 GmMADS4蛋白二级结构预测
大写字母表示GmMADS4蛋白氨基酸序列;小写字母代表不同的二级结构,h代表α螺旋,c代表无规则卷曲,e代表延伸链,t代表β转角
Figure 2. Secondary structure prediction of GmMADS4
Capital letters indicate amino acid sequence of GmMADS4 protein; Lowercase letters represent different secondary structures, where h stands for α helix, c for random coil, e for extended strand, and t for β turn
![]()
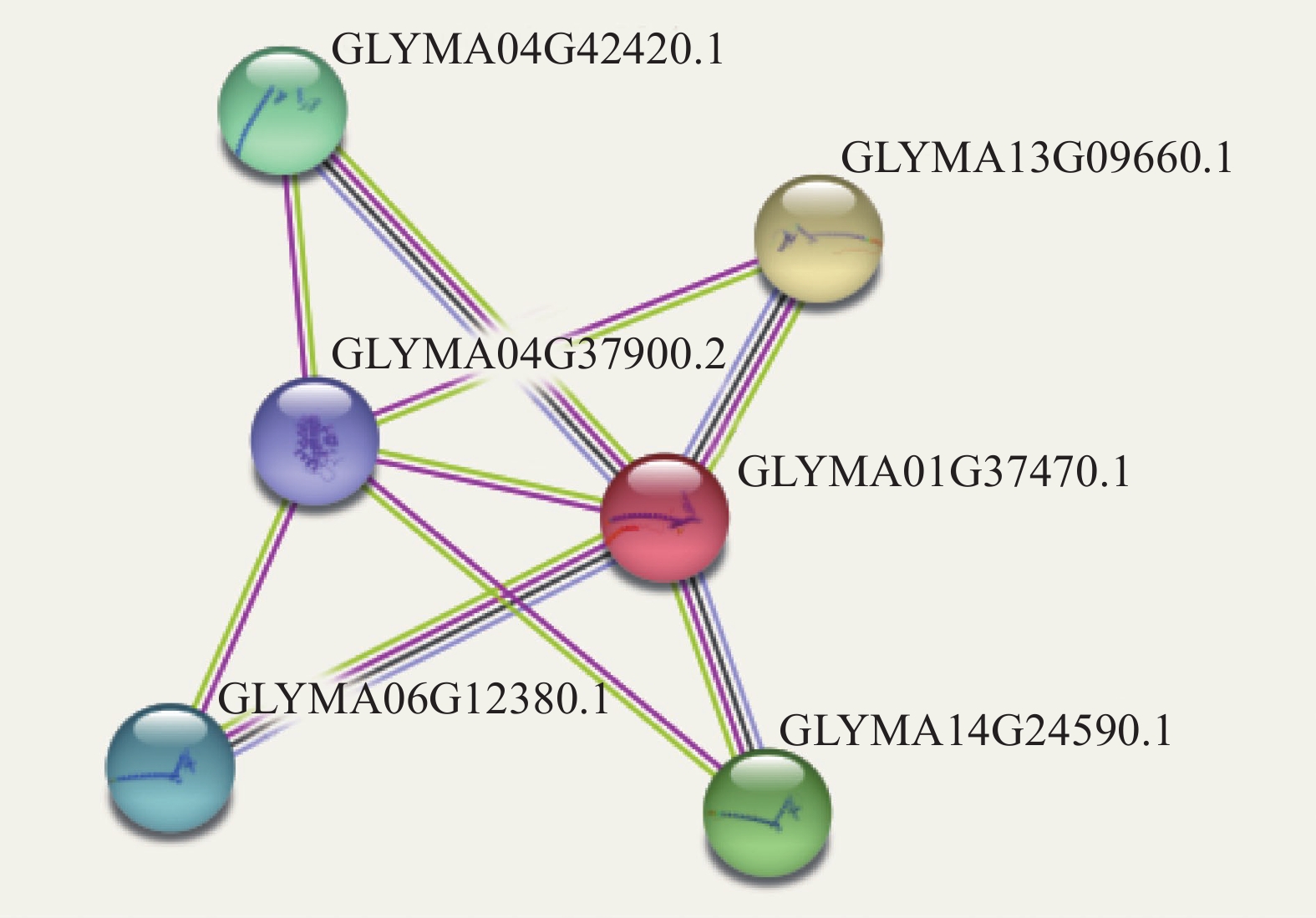
图 4 GmMADS4互作蛋白预测
不同颜色的圆球表示不同的蛋白,其中红色为GmMADS4;直线相连的2个蛋白预测存在相互作用
Figure 4. Interaction protein prediction of GmMADS4
Different colored spheres indicate different proteins, red is GmMADS4; Two proteins connected in a straight line are predicted to have an interaction
![]()
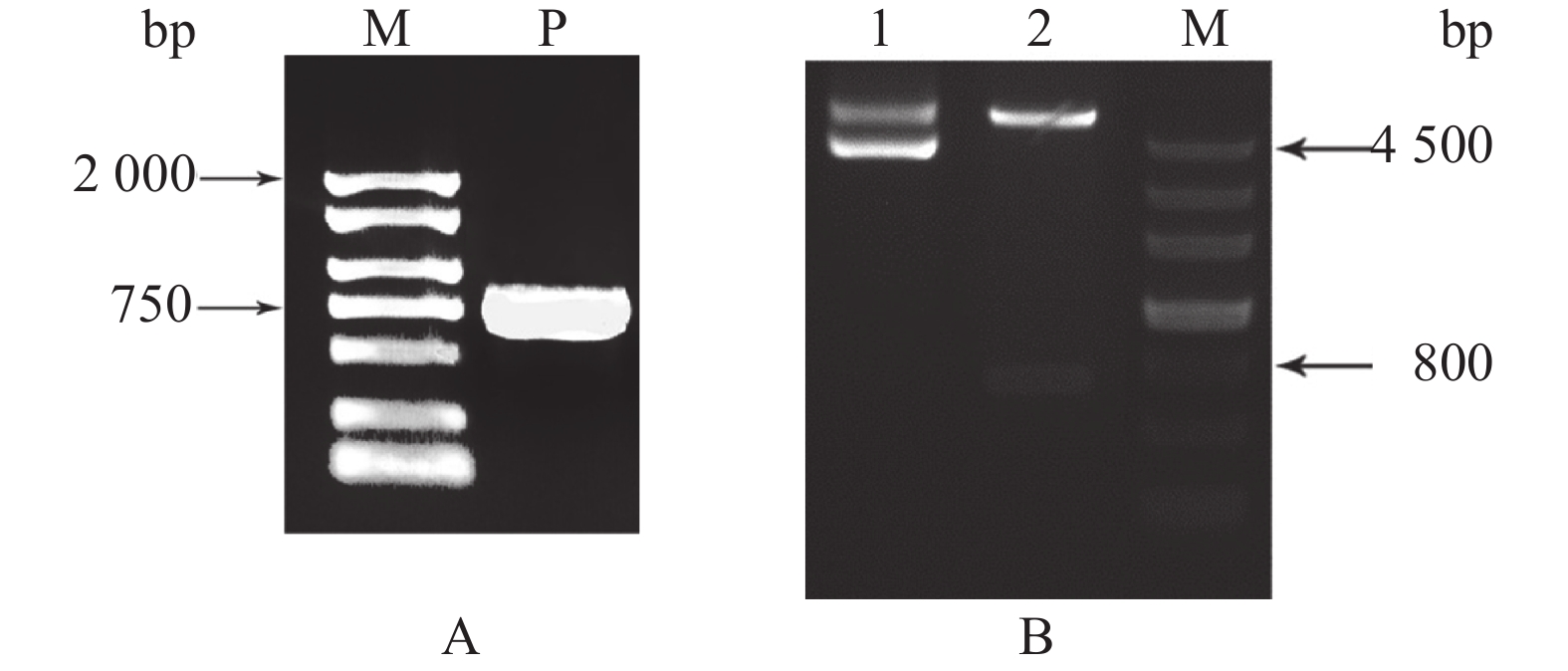
图 5 GmMADS4基因克隆及酶切检测
A:GmMADS4基因PCR扩增,B:GmMADS4-pTF101s重组质粒酶切检测;M代表DNA marker,P代表PCR产物,泳道1为重组质粒酶切前,泳道2为重组质粒酶切后
Figure 5. Cloning and enzyme digestion of GmMADS4
A: PCR amplification of GmMADS4 gene, B: Digestion verification of GmMADS4-pTF101s recombinant plasmid; M represents DNA marker, P represents PCR product, lane 1 is the recombinant plasmid before digestion, lane 2 is the recombinant plasmid after digestion
![]()
图 6 GmMADS4在大豆不同部位的表达分析
Figure 6. Expression pattern analysis of GmMADS4 in different soybean organs
![]()
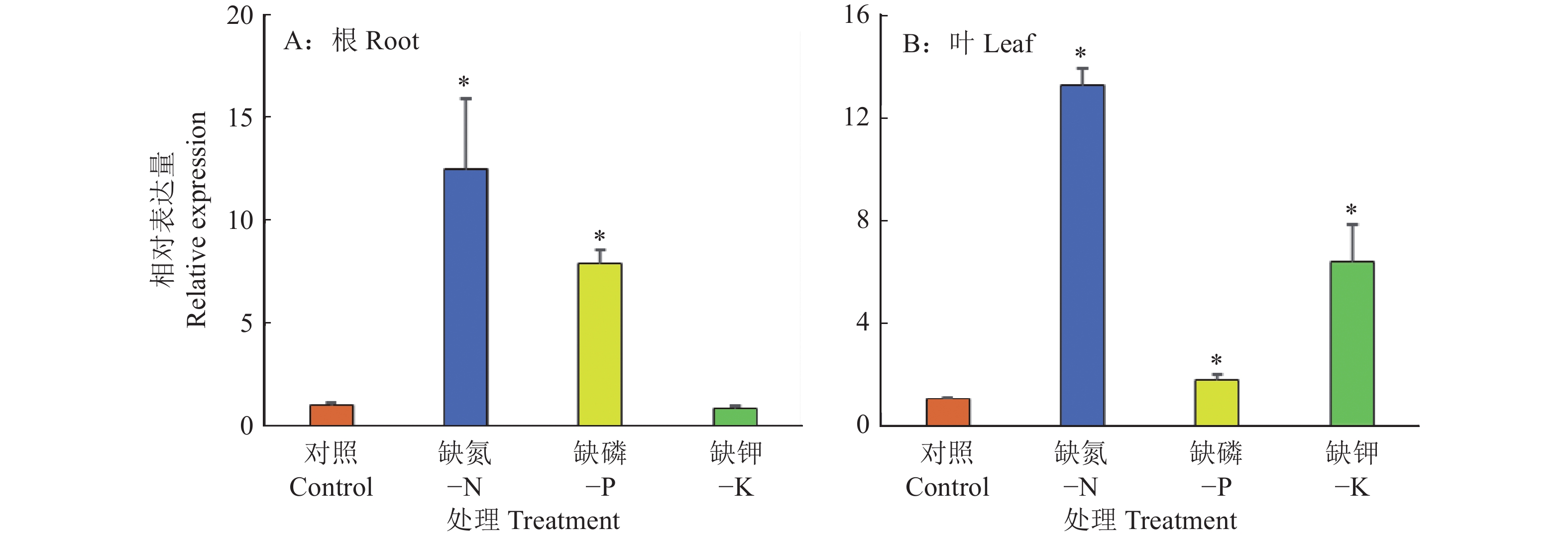
图 7 GmMADS4在大豆根和叶中响应缺素的表达模式分析
“*”表示缺素处理与对照相比差异显著(P < 0.05,t检验)
Figure 7. Expression pattern analysis of GmMADS4 in response to element deficiency in soybean roots and leaves
“*” indicates significant difference between element deficiency treatment and control (P < 0.05, t test)
![]()
图 9 超量表达GmMADS4转基因复合植株的生长表型
Figure 9. Growth phenotypes of overexpressing GmMADS4 transgenic composite plants
![]()
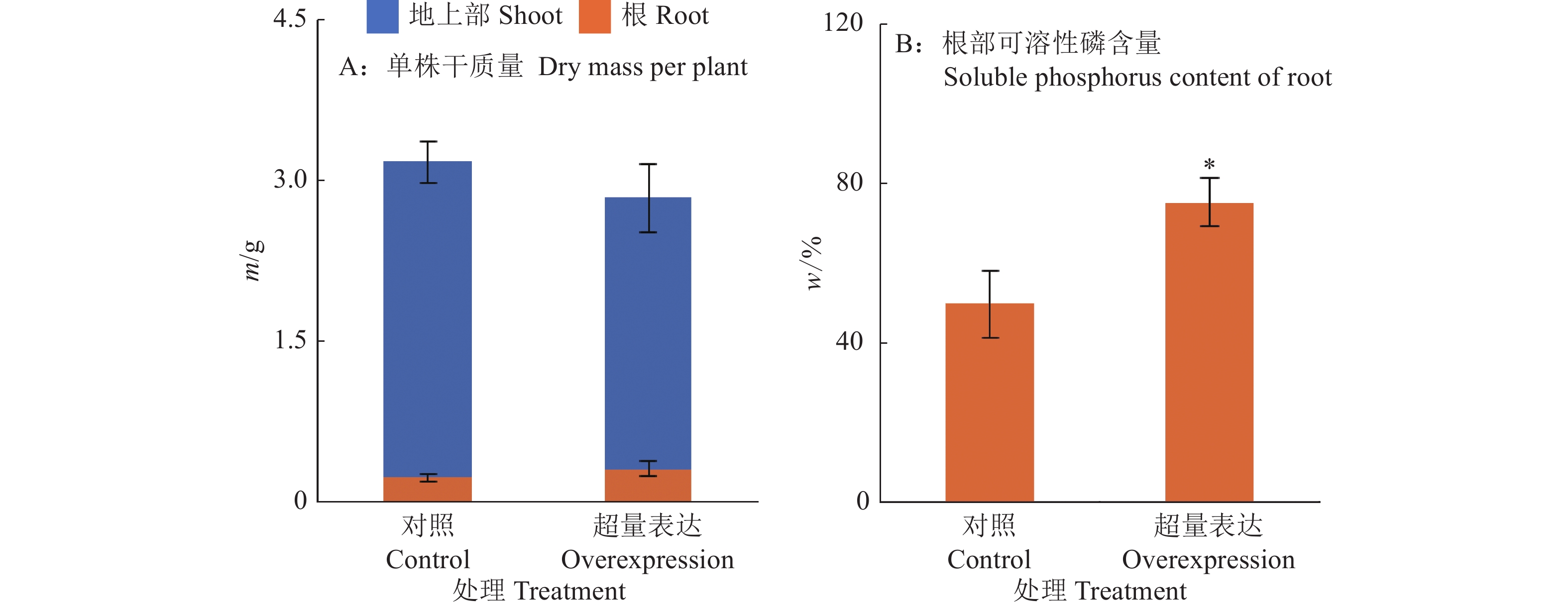
图 10 超量表达GmMADS4对大豆复合植株生物量和可溶性磷含量的影响
“*”表示对照和超量表达处理差异显著(P < 0.05,t检验)
Figure 10. Effects of overexpressing GmMADS4 on biomass and soluble phosphorus content of soybean composite plants
“*” indicates significant difference between control and overexpression treatment (P < 0.05, t test)
表 1 本研究所用到的引物
Table 1 Primers used in this study
引物名称 Primer name 引物序列(5′→3′)1)Primer sequence 注释 Annotation OE-GmMADS4-F C GAGCTCATGGGTCGTGGCAAGAT 超量表达 Overexpression OE-GmMADS4-R GC TCTAGATCAAGCAAGGCGAAGGTC GFP-GmMADS4-F GG GGTACCGATGGGTCGTGGCAAGAT 融合绿色荧光蛋白 Fused with GFP GFP-GmMADS4-R CG GGATCCCGAGCAAGGCGAAGGTCATG RT-GmMADS4-F GAGACAGATCAGGCATAGGA RT-qPCR RT-GmMADS4-R CCTACAGGTATCAGTCCGAG RT-GmEF-1α-F TGCAAAGGAGGCTGCTAACT RT-qPCR RT-GmEF-1α-R CAGCATCACCGTTCTTCAAA 1) 下划线为酶切位点 1) The restriction sites are underlined  下载: 导出CSV
下载: 导出CSV
-
[1] ALVAREZ-BUYLLA E R, PELAZ S, LILJEGREN S J, et al. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals[J]. Proceedings of the National Academy of Sciences of the United States of America, 2000, 97(10): 5328-5333. doi: 10.1073/pnas.97.10.5328
[2] BECKER A, THEISSEN G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants[J]. Molecular Phylogenetics and Evolution, 2003, 29(3): 464-489. doi: 10.1016/S1055-7903(03)00207-0
[3] 王莹, 穆艳霞, 王锦. MADS-box基因家族调控植物花器官发育研究进展[J]. 浙江农业学报, 2021, 33(6): 1149-1158. [4] PAUL P, DHATT B K, MILLER M, et al. MADS78 and MADS79 are essential regulators of early seed development in rice[J]. Plant Physiology, 2020, 182(2): 933-948. doi: 10.1104/pp.19.00917
[5] KANG I H, STEFFEN J G, PORTEREIKO M F, et al. The AGL62 MADS domain protein regulates cellularization during endosperm development in Arabidopsis[J]. The Plant Cell, 2008, 20(3): 635-647. doi: 10.1105/tpc.107.055137
[6] ZHANG G, XU N, CHEN H, et al. OsMADS25 regulates root system development via auxin signalling in rice[J]. The Plant Journal, 2018, 95(6): 1004-1022. doi: 10.1111/tpj.14007
[7] MA W Y, LIU W, HOU W S, et al. GmNMH7, a MADS-box transcription factor, inhibits root development and nodulation of soybean (Glycine max [L.] Merr.)[J]. Journal of Integrative Agriculture, 2019, 18(3): 553-562. doi: 10.1016/S2095-3119(18)61992-6
[8] 姚琦园, 李纷芬, 张林成, 等. 植物MADS-box转录因子参与调控非生物胁迫的研究进展[J]. 江西农业学报, 2018, 30(5): 73-79. doi: 10.19386/j.cnki.jxnyxb.2018.05.15 [9] ZHAO P X, ZHANG J, CHEN S Y, et al. Arabidopsis MADS-box factor AGL16 is a negative regulator of plant response to salt stress by downregulating salt-responsive genes[J]. New Phytologist, 2021, 232(6): 2418-2439. doi: 10.1111/nph.17760
[10] 闫凌月, 张豪健, 郑雨晴, 等. 转录因子OsMADS25提高水稻对低温的耐受性[J]. 遗传, 2021, 43(11): 1078-1087. doi: 10.16288/j.yczz.21-217 [11] ZHANG X, LI L, YANG C, et al. GsMAS1 encoding a MADS-box transcription factor enhances the tolerance to aluminum stress in Arabidopsis thaliana[J]. International Journal of Molecular Sciences, 2020, 21(6): 2004. doi: 10.3390/ijms21062004.
[12] YU C, SU S, XU Y, et al. The effects of fluctuations in the nutrient supply on the expression of five members of the AGL17 clade of MADS-box genes in rice[J]. PLoS One, 2014, 9(8): e105597. doi: 10.1371/journal.pone.0105597
[13] 曹永强, 王昌陵, 王文斌, 等. 国内外大豆产业、科技现状浅析与我国大豆产业发展思考[J]. 辽宁农业科学, 2019(6): 44-48. doi: 10.3969/j.issn.1002-1728.2019.06.011 [14] 田江, 梁翠月, 陆星, 等. 根系分泌物调控植物适应低磷胁迫的机制[J]. 华南农业大学学报, 2019, 40(5): 175-185. doi: 10.7671/j.issn.1001-411X.201905068 [15] 李欣欣, 杨永庆, 钟永嘉, 等. 豆科作物适应酸性土壤的养分高效根系遗传改良[J]. 华南农业大学学报, 2019, 40(5): 186-194. doi: 10.7671/j.issn.1001-411X.201905067 [16] 刘国选, 陈康, 陆星, 等. 大豆GmPIN2b调控根系响应低磷胁迫的功能研究[J]. 华南农业大学学报, 2021, 42(4): 33-41. doi: 10.7671/j.issn.1001-411X.202010014 [17] FAN C M, WANG X, WANG Y W, et al. Genome-wide expression analysis of soybean MADS genes showing potential function in the seed development[J]. PLoS One, 2013, 8(4): e62288. doi: 10.1371/journal.pone.0062288
[18] YAO Z, TIAN J, LIAO H. Comparative characterization of GmSPX members reveals that GmSPX3 is involved in phosphate homeostasis in soybean[J]. Annals of Botany, 2014, 114(3): 477-488. doi: 10.1093/aob/mcu147
[19] ZHU S, CHEN M, LIANG C, et al. Characterization of purple acid phosphatase family and functional analysis of GmPAP7a/7b involved in extracellular ATP utilization in soybean[J]. Frontiers in Plant Science, 2020, 11: 661. doi: 10.3389/fpls.2020.00661.
[20] LIU Y, XUE Y, XIE B, et al. Complex gene regulation between young and old soybean leaves in responses to manganese toxicity[J]. Plant Physiology and Biochemistry, 2020, 155: 231-242. doi: 10.1016/j.plaphy.2020.07.002
[21] ZHUANG Q, XUE Y, YAO Z, et al. Phosphate starvation responsive GmSPX5 mediates nodule growth through interaction with GmNF-YC4 in soybean (Glycine max)[J]. The Plant Journal, 2021, 108(5): 1422-1438. doi: 10.1111/tpj.15520
[22] MURPHY J, RILEY J P. A modifed single solution method for the determination of phosphate in natural water[J]. Analytica Chimica Acta, 1962, 27: 31-36. doi: 10.1016/S0003-2670(00)88444-5
[23] JACK T, BROCKMAN L L, MEYEROWITZ E M. The homeotic gene APETALA3 of Arabidopsis thaliana encodes a MADS box and is expressed in petals and stamens[J]. Cell, 1992, 68(4): 683-697. doi: 10.1016/0092-8674(92)90144-2
[24] PAR̆ENICOVÁ L, DE FOLTER S, KIEFFER M, et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis[J]. The Plant Cell, 2003, 15(7): 1538-1551. doi: 10.1105/tpc.011544
[25] SMACZNIAK C, IMMINK R G, MUINO J M, et al. Characterization of MADS-domain transcription factor complexes in Arabidopsis flower development[J]. Proceedings of the National Academy of Sciences of the United States of America, 2012, 109(5): 1560-1565. doi: 10.1073/pnas.1112871109
[26] NAN H, CAO D, ZHANG D, et al. GmFT2a and GmFT5a redundantly and differentially regulate flowering through interaction with and upregulation of the bZIP transcription factor GmFDL19 in soybean[J]. PLoS One, 2014, 9(5): e97669. doi: 10.1371/journal.pone.0097669
[27] CASTELAN-MUNOZ N, HERRERA J, CAJERO-SANCHEZ W, et al. MADS-box genes are key components of genetic regulatory networks involved in abiotic stress and plastic developmental responses in plants[J]. Frontiers in Plant Science, 2019, 10: 853. doi: 10.3389/fpls.2019.00853.
[28] GAN Y, FILLEUR S, RAHMAN A, et al. Nutritional regulation of ANR1 and other root-expressed MADS-box genes in Arabidopsis thaliana[J]. Planta, 2005, 222(4): 730-742. doi: 10.1007/s00425-005-0020-3
[29] GAN Y B, ZHOU Z J, AN L J, et al. A comparison between northern blotting and quantitative real-time PCR as a means of detecting the nutritional regulation of genes expressed in roots of Arabidopsis thaliana[J]. Agricultural Sciences in China, 2011, 10(3): 335-342. doi: 10.1016/S1671-2927(11)60012-6
[30] GAN Y, BERNREITER A, FILLEUR S, et al. Overexpressing the ANR1 MADS-box gene in transgenic plants provides new insights into its role in the nitrate regulation of root development[J]. Plant and Cell Physiology, 2012, 53(6): 1003-1016. doi: 10.1093/pcp/pcs050
[31] YAN Y, WANG H, HAMERA S, et al. MiR444a has multiple functions in the rice nitrate-signaling pathway[J]. The Plant Journal, 2014, 78(1): 44-55. doi: 10.1111/tpj.12446
[32] HUANG S, LIANG Z, CHEN S, et al. A transcription factor, OsMADS57, regulates long-distance nitrate transport and root elongation[J]. Plant Physiology, 2019, 180(2): 882-895. doi: 10.1104/pp.19.00142
[33] MA J, YANG Y, LUO W, et al. Genome-wide identification and analysis of the MADS-box gene family in bread wheat (Triticum aestivum L.)[J]. PLoS One, 2017, 12(7): e0181443. doi: 10.1371/journal.pone.0181443
-
期刊类型引用(1)
1. 解雅茹,金昊延,孔辰,蔡蓓,张令锴. CRISPR/Cas9系统在家畜生殖细胞中的研究进展. 畜牧兽医学报. 2025(02): 479-491 .  百度学术
百度学术
其他类型引用(0)














计量
- 文章访问数: 204
- HTML全文浏览量: 24
- PDF下载量: 39
- 被引次数: 1
