
Distribution of ‘Candidatus Liberibacter asiaticus’ in the fruit branches of four citrus cultivars
-
摘要:目的
亚洲种韧皮部杆菌(‘Candidatus Liberibacter asiaticus’, CLas)是引起柑橘黄龙病(Citrus Huanglongbing, HLB)的优势种,也是引发我国柑橘HLB的唯一病原菌。探索CLas在柑橘果枝内的时空分布规律,有利于及时准确地对其进行检测,对HLB的防控具有重要意义。
方法以砂糖橘、W−默科特、红心蜜柚和沙田柚不同生长期的果枝为材料,利用实时荧光定量PCR检测果枝CLas含量,研究其时空分布规律。
结果CLas在病枝中分布不均匀,在果实处(橘络、果中轴或种皮)富集。对于宽皮柑橘的砂糖橘和杂柑的W−默科特,成熟期果实橘络中的CLas含量最高;而对于柚类的红心蜜柚和沙田柚,成熟期果轴中的CLas含量最高。砂糖橘果实膨大与成熟过程中,CLas随着营养物质的流动从源器官(叶片)向库器官(果实)分流并聚集;果轴和橘络中的CLas含量不断增加,且橘络中的CLas含量显著上升。
结论CLas随着柑橘果实生长和成熟而汇集,结合源库理论可解析CLas在果枝上的运转和分布规律。
Abstract:Objective‘Candidatus Liberibacter asiaticus’ (CLas), the main species causing Citrus Huanglongbing (HLB), is also the sole pathogen causing agent of HLB in China. Exploring the temporal and spatial distribution of CLas in citrus fruit branch would help timely and accurate detection of CLas, and is of great significance to the prevention and control of HLB.
MethodThe titers of CLas were measured in HLB-affected fruit branches of four traditional citrus cultivars namely Citrus reticulata Blanco cv. Shatangju, (C. reticulata × C. sinensis) cv. W-Murcott, C. maxima cv. Hongxinmi Yu and C. maxima cv. Shatian Yu using real time PCR method. The temporal and spatial distribution of CLas in citrus fruit branches were studied.
ResultCLas was unevenly distributed in fruit branches with enrichment in the fruits (Fruit pith, centrol axis or seed coat). For the mandarin of Shatangju and W-Murcott, CLas titers were the highest in the piths at the fruit mature stage. For the pomelos of Hongxinmi Yu and Shatian Yu, CLas titers were the highest in the fruit central axes at ripe stage. CLas transferred from the source organs (leaves) to the sink organs (fruits) along with the flow of nutrients in the processes of fruit swelling and ripening. CLas titers kept increasing in central axes and fruit pith, especially significantly increased in fruit pith.
ConclusionCLas is enriched with the fruit developmental stages, and such transfer and distribution pattern of CLas in fruit branches can be explained by the theory of plant source-sink relationship.
-
猪是重要的农业动物,其重要经济性状的遗传解析对于提高猪生产效率至关重要。目前,全基因组关联分析 (Genome-wide association studies, GWAS)已成为挖掘畜禽性状重要分子标记和功能基因的主要手段之一[1],人们使用该方法陆续进行了猪乳头数[2]、背膘厚度[3-4]、内脏质量[5]、产仔数[6]等重要生长、繁殖性状的关键位点分析。随着多组学技术普及以及猪基因型−组织表达(Pig genotype-tissue expression, PigGTEx)项目的开展[7],我们能够将基因组和转录组等多组学数据进行联合分析,找到与分子表型相关的数量性状位点[8],有助于解析猪复杂性状的分子遗传机制。
作为GWAS后分析的重要方法,全转录组关联研究(Transcriptome-wide association study, TWAS)被提出后得到了广泛应用。TWAS能将基因表达、GWAS检测出的显著变异以及性状关联起来。TWAS分析的关键是在参考群体中构建基因表达的预测模型,预测GWAS群体中的个体基因表达水平[9]。提高TWAS预测模型的准确性有助于提升TWAS分析的统计效力。在现有的TWAS软件中,PrediXcan软件使用弹性网络的线性回归模型拟合基因顺式单核苷酸多态性(Single nucleotide polymorphism, SNP)以预测组织中的基因表达水平[10],Fusion软件使用贝叶斯稀疏线性混合模型(Bayesian sparse linear mixed model, BSLMM)预测组织中的基因表达水平[11]。PigGTEx项目使用S-PrediXcan和S-MultiXcan中的嵌套交叉验证弹性网络模型,利用基因的顺式SNP(cis-SNP)预测表达量并用于后续TWAS分析[7]。然而,目前在猪等畜禽群体中仍缺少对基因表达量预测模型的效果评估。
在本研究中,我们以猪群体数据为对象,比较多种机器学习回归模型(包括线性回归模型和非线性回归模型)预测基因表达量的效果,并且探究基因cis-SNP数量和基因顺式遗传力(cis-heritability, cis-h2)对预测准确性的影响,为后续TWAS模型选择和优化提供参考。
1. 材料与方法
1.1 试验数据
本研究选取的
1321 头猪的肌肉组织基因型数据和基因表达量数据均来自PigGTEx项目(https://piggtex.farmgtex.org)。基因型数据只保留小等位基因频率(Minor allele frequency, MAF)> 0.05的SNP位点,质控后保留了2928814 个SNP。猪肌肉组织基因表达数据中共包含13454 个基因的TPM(Transcripts per million)表达量矩阵。使用R包edgeR[12]中的TMM(Trimmed mean of M-value)方法对TPM标准化,并进行逆正态转换(Inverse normal transformation, INT)。后续分析均使用INT的TMM基因表达矩阵。为了校正数据中的群体分层,我们使用R包SNPRelate v1.26.0[13]计算基因型主成分,选取前10个基因型主成分作为协变量。为了消除基因表达数据的潜在混杂效应,首先使用PEER(Probabilistic estimation of expression residual)方法[14]估计基因表达量数据中潜在的混杂因素,最终选择前10个PEER因子作为协变量。本研究利用一般线性回归模型校正表达量数据中的群体分层和潜在混杂因素,模型为:
$$ \boldsymbol{y}={\boldsymbol{X}}_{\bf{cov}}\boldsymbol{\beta }+\boldsymbol{\varepsilon } \text{,} $$ (1) 式中,
$ \boldsymbol{y} $ 是基因表达量向量,$ {\boldsymbol{X}}_{\bf{cov}} $ 是协变量矩阵,$ \boldsymbol{\beta } $ 是回归系数向量,$ \boldsymbol{\varepsilon } $ 是误差项向量。我们使用上述计算得到的10个基因型主成分和10个PEER因子构建协变量矩阵,对于第$ i $ 个基因,使用最小二乘法估计回归系数$ {\hat {\boldsymbol{\beta }}}_{\boldsymbol{i}} $ ,再计算校正后的基因表达量($ {\boldsymbol{y}}_{\text{c}} $ ):$$ {\boldsymbol{y}}_{\text{c}}=\boldsymbol{y}-{\boldsymbol{X}}_{{\bf{cov}}}\hat {\boldsymbol{\beta }_{\boldsymbol{i}}} {\text{。}} $$ (2) 本研究依据Ensembl数据库中的猪基因注释文件(Sscrofa 11.1 v100),选取猪1号染色体上的
1091 个蛋白编码基因用于后续分析。1.2 基因表达量的机器学习预测模型
本研究使用的机器学习回归模型均来自Python v3.12.3中的scikit-learn包,总共18种监督学习模型,可分为最小二乘法回归模型、正则化回归模型、岭(Ridge)回归模型、支持向量机回归模型、广义线性模型、K最近邻回归(K-nearest neighbors regression, KNN)模型和树回归模型7类。
1.2.1 最小二乘法回归模型
最小二乘法回归模型包括普通最小二乘法回归(Ordinary least squares, OLS)和偏最小二乘法回归(Partial least squares, PLS)模型,是线性回归模型,其基本模型如下:
$$ \boldsymbol{y}=\boldsymbol{X}\boldsymbol{\beta }+\boldsymbol{\varepsilon } \text{,} $$ (3) 式中,
$ \boldsymbol{X} $ 是个体基因型矩阵,$ \boldsymbol{\beta } $ 是cis-SNP效应值向量。假设$ {{\boldsymbol{y}}}_{{\boldsymbol{1}}},{{\boldsymbol{y}}}_{{\boldsymbol{2}}},\cdots ,{{\boldsymbol{y}}}_{{\boldsymbol{n}}} $ 为$ n $ 个样本在特定组织中的基因表达量,则效应值向量求解公式(优化函数)如下:$$ \hat {\boldsymbol{\beta }}={\mathrm{arg}}\underset{{\boldsymbol{\beta}} }{\mathrm{min}}\left(1/2\right)\displaystyle\sum\nolimits _{i=1}^{n}{\left({{\boldsymbol{y}}}_{{\boldsymbol{i}}}-{{{\boldsymbol{x}}}_{{\boldsymbol{i}}}}^{{\mathrm{T}}}\boldsymbol{\beta }\right)}^{2} {\text{,}} $$ (4) 式中,
${\boldsymbol{y}}_{\boldsymbol{i}} $ 和${\boldsymbol{x}}_{\boldsymbol{i}} $ 分别是第i个样本的基因表达量和cis-SNP向量。PLS同时考虑因变量和自变量之间的协方差求解回归系数,虽然计算更复杂,但在处理高维和共线性数据时能够提供比OLS更稳健的模型[15]。1.2.2 正则化回归模型
正则化回归模型包括岭回归、Lasso回归、LassoLars回归和弹性网络(Elastic net)回归。这些模型通过在OLS基础上引入罚函数(即正则化项)来防止过拟合,提高模型的泛化能力,是线性模型,其优化函数如下:
$$ \begin{split} \hat {\boldsymbol{\beta }}=&\;{\mathrm{arg}}\underset{{\boldsymbol{\beta}} }{\mathrm{min}}\left(1/2\right)\displaystyle\sum\nolimits _{i=1}^{n}{\left({{\boldsymbol{y}}}_{{\boldsymbol{i}}}-{{{\boldsymbol{x}}}_{{\boldsymbol{i}}}}^{{\mathrm{T}}}\boldsymbol{\beta }\right)}^{2}+\\&\lambda \left[\left(1-\alpha \right){\|\boldsymbol{\beta }\|}_{2}^{2}+\alpha {\|\boldsymbol{\beta }\|}_{1}\right] \text{,} \end{split} $$ (5) 式中,
$ {\|\boldsymbol{\beta }\|}_{1} $ 和$ {\|\boldsymbol{\beta }\|}_{2} $ 分别是$ \boldsymbol{\beta } $ 的L1范数(即$ \boldsymbol{\beta } $ 系数的绝对值和)和L2范数(即$ \boldsymbol{\beta } $ 系数的平方和),$ \lambda $ 是正则化强度参数。Ridge回归只包含L2范数[16],即式(5)中L1和L2正则化的权重比例系数
$ \alpha =0 $ 。Lasso回归和LassoLars回归只包含L1范数,即$ \alpha =1.0 $ ,两者优化函数相同,由于LassoLars回归是一种逐步回归方法,每一步选择1个变量进入模型,LassoLars回归会在每一步中使用L1正则化调整回归系数,与Lasso回归相比能更高效生成稀疏解,适用于高维数据[17]。Elastic net回归包含L1和L2范数,可以调整$ \alpha $ 使模型同时具备稀疏性和稳定性[18],在本研究中取$ \alpha =0.5 $ ,即L1和L2正则化权重相同。Lasso、LassoLars和Elastic net回归受$ \lambda $ 影响大,且存在收敛问题,即无法从已有数据中预测出结果。经前期验证,针对本研究的数据,我们在使用这3种回归模型时,先对每个基因表达量预测进行5折交叉验证(将数据集分为5个子集,循环使用每个子集作为验证集,其余子集作为训练集),从$ 0.100 $ 、$ 0.050 $ 、$0.010 $ 、$ 0.005 $ 、$ 0.001 $ 中选择对每个基因预测效果最好的$ \lambda $ 取值,用于后续分析。1.2.3 岭回归模型
岭回归模型包括核岭(Kernel ridge)回归和贝叶斯岭(Bayesian ridge)回归模型。Kernel ridge在OLS基础上引入了岭正则化和核函数,其优化函数如下:
$$ \hat {\boldsymbol{\beta }}={\mathrm{arg}}\underset{{\boldsymbol{\beta}} }{\mathrm{min}}\left(1/2\right)\displaystyle\sum\nolimits _{i=1}^{n}{\left({{\boldsymbol{y}}}_{{\boldsymbol{i}}}-\boldsymbol{K}\boldsymbol{\beta }\right)}^{2}+\lambda {\|\boldsymbol{\beta }\|}^{2} \text{,} $$ (6) 式中,
$ \boldsymbol{K} $ 是$ n\times n $ 的核矩阵。最开始核函数在支持向量机(Support vector machine, SVM)中使用,可以大大降低其非线性变换的计算量,之后进一步将核函数引入到传统机器学习算法中,能方便地把线性算法转换为非线性算法[19]。其中,核函数有4种。线性核(Linear kernel)形式为:
$$ K\left({{\boldsymbol{x}}}_{{\boldsymbol{i}}},{{\boldsymbol{x}}}_{{\boldsymbol{j}}}\right)={{\boldsymbol{x}}}_{{\boldsymbol{i}}} {{\boldsymbol{x}}}_{{\boldsymbol{j}}}{\text{,}}$$ (7) 式中,
${{\boldsymbol{x}}}_{{\boldsymbol{j}}} $ 是第j个样本的cis-SNP向量,适用于线性数据或特征数远大于样本数的数据。多项式核(Polynomial kernel,后文简称为poly)形式为:
$$ K\left({{\boldsymbol{x}}}_{{\boldsymbol{i}}},{{\boldsymbol{x}}}_{{\boldsymbol{j}}}\right)={\left({{\boldsymbol{x}}}_{{\boldsymbol{i}}} {{\boldsymbol{x}}}_{{\boldsymbol{j}}}+\tau \right)}^{d}{\text{,}}$$ (8) 式中,
$ \tau $ 是常数,$ d $ 是多项式的度数,适用于存在非线性关系的数据。径向基函数核 (RBF kernel)形式为:
$$ K\left({{\boldsymbol{x}}}_{{\boldsymbol{i}}},{{\boldsymbol{x}}}_{{\boldsymbol{j}}}\right)={\mathrm{exp}}\left(-\dfrac{{\|{{\boldsymbol{x}}}_{{\boldsymbol{i}}}-{{\boldsymbol{x}}}_{{\boldsymbol{j}}}\|}^{2}}{2{\sigma }^{2}}\right){\text{,}}$$ (9) 式中,
$ \sigma $ 是核函数参数,同样适用于存在非线性关系的数据。Sigmoid核 (Sigmoid kernel)形式为:
$$ K\left({{\boldsymbol{x}}}_{{\boldsymbol{i}}},{{\boldsymbol{x}}}_{{\boldsymbol{j}}}\right)=\mathrm{tanh}\left[\delta \left({{\boldsymbol{x}}}_{{\boldsymbol{i}}} {{\boldsymbol{x}}}_{{\boldsymbol{j}}}\right)+c\right]{\text{,}}$$ (10) 式中,
$ \delta $ 和$ c $ 是可调参数,通常用于神经网络的训练。Bayesian ridge与传统岭回归相比通过考虑后验分布来量化模型系数的不确定性。
1.2.4 支持向量机回归模型
支持向量机回归模型(Support vector regression, SVR)的目标是找到一个函数
$ f\left({\boldsymbol{x}}\right)=\left\langle{\boldsymbol{\beta },{\boldsymbol{x}}}\right\rangle+b $ ,使所有$ \left({{\boldsymbol{x}}}_{{\boldsymbol{i}}},{{\boldsymbol{y}}}_{{\boldsymbol{i}}}\right) $ 的误差$ \left|{{\boldsymbol{y}}}_{{\boldsymbol{i}}}-f\left({{\boldsymbol{x}}}_{{\boldsymbol{i}}}\right)\right| $ 大部分位于给定的$ {\boldsymbol{\varepsilon}} $ 内,目标优化函数如下:$$ \hat {\boldsymbol{\beta }}={\mathrm{arg}}\underset{{\boldsymbol{\beta}} ,b}{\mathrm{min}}\left(1/2\right){\|\boldsymbol{\beta }\|}^{2}+{\mathrm{loss}} \text{,} $$ (11) 式中,loss为损失函数。
与Kernel ridge回归类似,SVR也可引入核函数,本研究的SVR使用linear、poly和RBF这3种核函数构建预测模型。SVR适用于具有复杂分布的数据或者高维数据。
1.2.5 广义线性模型
广义线性模型(Generalize linear model, GLM) 主要由3个部分组成:随机部分
$ {Y}_{1},{Y}_{2},\cdots ,{Y}_{n} $ 服从的分布来自指数分布族;系统部分由线性预测器组成;存在一个单调可微的连接函数,用于连接随机部分的期望和系统部分的线性预测值,即:$$ g\left(\mu \right)=\eta \text{,} $$ (12) 式中,
$ \mu $ 是$ \mathit{Y} $ 的期望,$ \eta $ 是线性预测器。GLM存在多种连接函数,受
$ Y $ 的分布决定。本研究中,我们假定$ Y $ 的分布属于正态分布,则连接函数为$ \mu =\eta $ 。GLM适用于处理误差向量存在非恒等方差的情况,也适用于存在非线性关系的数据。1.2.6 K最近邻回归模型
KNN是一种非参数方法,无需数据分布假设,直接基于实例预测。其流程为:针对待测样本,基于欧氏距离找出训练集中的K个最近邻,取其基因表达量均值作为预测值。该方法无需训练、对异常值不敏感且适应非线性关系,但高维数据下性能显著下降。
1.2.7 树回归模型
树回归包括决策树回归(Decision tree regression)和随机森林回归(Random forest regression),是非参数回归方法。决策树递归地将基因型矩阵
$ {\boldsymbol{X}}$ 分割成更小的子集来构建树结构,通过最小化基因表达量$ {\boldsymbol{y}} $ 均方误差选择分割点$ s $ ,对于分割出的第$ j $ 个基因型矩阵(${\boldsymbol{X}}_{\boldsymbol{j}} $ )和选择的分割点$ s $ ,可以将基因型数据分成如下2个部分:$$ {K}_{1}\left(j,s\right)=\left\{\left({\boldsymbol{X}},{\boldsymbol{y}}\right)|{{\boldsymbol{X}}}_{{\boldsymbol{j}}}\in s\right\}{\text{,}} $$ (13) $$ {K}_{2}\left(j,s\right)=\left\{\left({\boldsymbol{X}},{\boldsymbol{y}}\right)|{{\boldsymbol{X}}}_{{\boldsymbol{j}}}\notin s\right\}{\text{,}}$$ (14) 式中,
$ K $ 是叶节点包含的样本集合。对每个子集递归使用类似的分割过程,直至达到数的最大深度。对于待预测基因表达量的个体基因型向量
$ {{\boldsymbol{x}}}_{{\boldsymbol{i}}} $ ,沿决策树从根节点基于分割点决策,最终到达1个叶节点,计算基因表达量均值用于预测样本的基因表达量,即:$$ \hat {{\boldsymbol{y}}}=\dfrac{1}{\left|K\right|}\displaystyle\sum _{i\in K}{{\boldsymbol{y}}}_{{\boldsymbol{i}}} 。 $$ (15) 随机森林回归通过集成多棵决策树进行预测,该模型虽结构复杂但假设条件少,默认参数通常接近最优[20]。通过集成策略提升了稳定性和预测精度,随机森林回归能有效缓解单棵树的过拟合问题,并能捕捉基因型特征与表达量间的复杂非线性关系。
1.3 模型的预测准确性评估
对于每个基因,选取转录起始位点(Transcription start site, TSS)上、下游1 Mb范围内的cis-SNP用于训练,在
1321 个样本中进行10折交叉验证并重复5次。在1次10折交叉验证中,将1321 个样本平均分成10份,循环选取其中的1份(132个样本)作为验证集,剩余9份(1189 个样本)作为训练集。我们通过计算每个验证集中真实基因表达量和模型预测基因表达量之间的皮尔逊相关系数的平方(R2)来衡量模型预测基因表达量的准确性。1.4 基因顺式遗传力计算
为探究基因遗传力与模型预测准确性的关系,选取每个基因TSS上、下游1 Mb范围内的cis-SNP,建立混合线性模型估算基因的cis-h2,统计模型为:
$$ \boldsymbol{y}={\boldsymbol{X}}_{\bf{c}\bf{o}\bf{v}}\boldsymbol{\beta }+\boldsymbol{g}+\boldsymbol{\varepsilon } \text{,} $$ (16) 式中,
${\boldsymbol{g}} $ 是与cis-SNP相关的随机效应,$ \boldsymbol{g}\sim N\left(0,{\sigma }_{g}^{2}\boldsymbol{G}\right) $ ,服从均值为0、协方差为${\sigma }_{g}^{2}\boldsymbol{G} $ 的多元正态分布,${\boldsymbol{G}} $ 是基因组遗传关系矩阵(Genetic relationship matrix, GRM),$ {\sigma }_{g}^{2}$ 是遗传方差。该分析基于PLINK v1.90提取每个基因的cis-SNP,利用GCTA v1.94.1软件[21]计算GRM,将其作为其随机效应,并将基因表达量作为表型,使用AIREML(Average information restricted maximum likelihood)估算基因的cis-h2。1.5 基因分组与筛选
为了研究基因cis-h2与模型预测准确性的关系,将基因按照cis-h2大小分成cis-h2≤0.01、0.01 <cis-h2≤0.10、0.10<cis-h2≤0.20、cis-h2>0.20这4个组,计算每组基因中不同模型的R2平均值。为了研究基因的cis-SNP数量对模型预测准确性的影响,计算所有基因cis-SNP数量的上、下四分位数和中位数,分别选取cis-SNP数量在[0,下四分位数]、(下四分位数,中位数]、(中位数,上四分位数]、大于上四分位数这4个范围内的基因,计算它们在不同模型中的R
2与cis-h2的皮尔逊相关系数(r)。 2. 结果与分析
2.1 不同模型预测基因表达量总体准确性
不同机器学习模型的总体预测效果如图1所示。Elastic net和Lasso 模型的R2平均值最高,分别为
0.0362 和0.0358 ;使用RBF非线性核的SVR和Kernel ridge的R2平均值次之,分别为0.0354 和0.0347 ;GLM_Gaussian和OLS模型的R2平均值最低,分别为0.0089 和0.0072 。所有模型中98%以上的基因R2<0.2,2%以内的基因R2>0.2。![]() 图 1 不同机器学习模型的总体预测准确性(R2)柱子中的红色圆点表示R2平均值,图中数字为平均值具体数值。Figure 1. Overall prediction accuracy (R2) of different machine learning modelsRed points in the columns indicate means of R2, the numbers in the figure are the values of means.
图 1 不同机器学习模型的总体预测准确性(R2)柱子中的红色圆点表示R2平均值,图中数字为平均值具体数值。Figure 1. Overall prediction accuracy (R2) of different machine learning modelsRed points in the columns indicate means of R2, the numbers in the figure are the values of means.2.2 不同模型预测准确性与基因顺式遗传力关系
18种机器学习模型预测准确性(R2)与基因cis-h2的关系如图2所示。在所有模型中,机器学习模型的R2与基因cis-h2呈正相关,两者间的皮尔逊相关系数大于0.60;对于整体R2平均值较高的模型,皮尔逊相关系数大于0.75。
![]() 图 2 不同机器学习模型预测准确性(R2)与基因顺式遗传力(cis-h2)的关系Figure 2. Relationship between the prediction accuracy (R2) of different machine learning models and the cis-heritability (cis-h2) of genes
图 2 不同机器学习模型预测准确性(R2)与基因顺式遗传力(cis-h2)的关系Figure 2. Relationship between the prediction accuracy (R2) of different machine learning models and the cis-heritability (cis-h2) of genes我们计算出所有基因的cis-h2中位数为
0.0267 ,平均值为0.0762 。基因按照cis-h2大小分组情况如表1所示。其中,cis-h2≤0.01的基因占全部基因的39.54%,cis-h2>0.20的基因占全部基因的12.88%。表 1 4组基因的顺式遗传力Table 1. Cis-heritability of four groups of genes顺式遗传力
cis-h2基因个数
Number of genes中位数
Median value平均值
Mean value标准误
Standard error≤0.01 433 1.000×10−6 1.000×10−3 1.02×10−4 (0.01, 0.10] 372 0.043 0.048 1.35×10−3 (0.10, 0.20] 139 0.139 0.144 2.49×10−3 > 0.20 141 0.281 0.315 9.21×10−3 选取每组基因预测R2平均值排名前7的机器学习模型,结果如图3所示。对于cis-h2≤0.01的基因,Lasso和Random forest的预测准确性最高,R2平均值分别为
0.0152 和0.0146 ;对于cis-h2范围在(0.01, 0.10]的基因,Elastic net和Lasso这2个正则化回归模型的预测准确性最高,R2平均值分别为0.0275 和0.0272 ;对于cis-h2范围在(0.10, 0.20]的基因,使用RBF非线性核的Kernel ridge和SVR的预测准确性最高,R2平均值分别为0.0530 和0.0526 ;对于cis-h2>0.20的基因,Elastic net和Lasso的预测准确性最高,R2平均值分别为0.115 0和0.107 0。![]() 图 3 不同顺式遗传力范围内排名前7的模型的预测准确性(R2)Figure 3. Prediction accuracy (R2) of the top 7 models within different cis-heritability ranges
图 3 不同顺式遗传力范围内排名前7的模型的预测准确性(R2)Figure 3. Prediction accuracy (R2) of the top 7 models within different cis-heritability ranges2.3 顺式SNP数量对不同模型预测准确性的影响
不同机器学习回归模型的算法存在差异,不同的特征数量(即cis-SNP数量)会对预测准确性有影响,一般情况模型预测准确性与cis-SNP数量呈正相关。计算得出cis-SNP数量与cis-h2的皮尔逊相关系数为0.236(P = 3.05×10−15)。基因cis-SNP数量与cis-h2存在相关性,同样表明cis-SNP数量对模型预测准确性有一定影响。
计算得到基因cis-SNP数量的下四分位数是
1564 ,中位数是3097 ,上四分位数是5384 。选取cis-SNP数量在[0,1564 ]、(1564 ,3097 ]、(3097 ,5384 ]、>5384 范围的基因,计算它们在不同模型中的R2与cis-h2的皮尔逊相关系数,得到结果如表2所示。除了OLS、Ridge、Kernel ridge_sigmoid、GLM_Gaussian 4个模型,其余14种模型在基因cis-SNP数量为(3097 ,5384 ]范围时预测准确性最高,说明cis-SNP数量过少或过多会使这些模型训练过程产生欠拟合或过拟合现象。OLS和GLM_Gaussian随基因cis-SNP数量增多,预测准确性提升,与基因cis-SNP数量为[0,1564 ]范围内相比,基因cis-SNP数量大于5384 时2个模型预测准确性分别提升8.78个百分点和10.87个百分点。表 2 不同顺式SNP数量范围的模型预测准确性(R2)与基因顺式遗传力(cis-h2)的相关系数Table 2. Correlation coefficient between the model prediction accuracy (R2) within different ranges of cis-SNP number and cis-heritability (cis-h2) of genes模型 Model 顺式SNP数量范围 The range of cis-SNP number [0, 1564 ]( 1564 ,3097 ]( 3097 ,5384 ]> 5384 OLS 0.638 0.647 0.673 0.694 PLS 0.656 0.668 0.698 0.647 Ridge 0.649 0.621 0.639 0.626 Lasso 0.782 0.780 0.795 0.774 LassoLars 0.796 0.792 0.801 0.767 Elastic net 0.806 0.802 0.811 0.792 Kernel ridge_linear 0.651 0.613 0.654 0.625 Kernel ridge_poly 0.787 0.803 0.814 0.808 Kernel ridge_RBF 0.794 0.805 0.810 0.768 Kernel ridge_sigmoid 0.657 0.650 0.653 0.615 Bayesian ridge 0.777 0.778 0.786 0.750 SVR_linear 0.617 0.617 0.634 0.632 SVR_poly 0.742 0.770 0.788 0.774 SVR_RBF 0.795 0.801 0.814 0.772 GLM_Gaussian 0.640 0.671 0.704 0.709 KNN 0.710 0.723 0.754 0.732 Decision tree 0.618 0.644 0.680 0.631 Random forest 0.756 0.766 0.783 0.773 3. 讨论与结论
个体的基因表达水平可以被分解为由遗传决定的成分(Genetically regulated expression,GReX)、由性状本身改变的成分和归因于环境和其他因素的剩余成分。对于大多数基因,其表达量被认为具有稀疏结构,即由几个主要SNP调控,每个SNP都对基因表达量有较大影响[22],而剩余的SNP对基因表达量都有微小的影响。利用SNP预测基因表达量是一个多元回归任务,包含多个SNP作为特征,预测准确性会直接影响TWAS后续进行基因表达量与表型之间的关联分析。经过前期研究发现,若使用所有SNP而非使用基因的cis-SNP进行基因表达量预测会产生较多噪声且运行速度缓慢[10],所以本研究使用了18种不同机器学习回归模型利用cis-SNP预测猪基因表达量。
结果表明,不同模型预测准确性(R2)与基因cis-h2呈正相关,但R2一般不高于cis-h2。使用机器学习回归模型预测的基因表达量实际上是预测基因表达水平的GReX,无法预测环境及其他因素对基因表达水平的影响。因此,基因的遗传力越大,即基因表达受遗传因素影响较大,模型的预测准确性越高。在本研究中,所有基因cis-h2的平均值为
0.0762 ,所有模型整体R2的平均值范围为0.0072 ~0.0362 ,符合预期结果。整体预测准确性最高的模型是Elastic net回归模型和Lasso回归模型,这与Gamazon等[10]的研究结果一致。Elastic net回归模型结合了Lasso回归的L1正则化项和Ridge回归的L2正则化项,通过灵活调整L1和L2正则化的权重比例系数
$ \alpha $ ,可以防止模型过拟合,且有利于生成稀疏模型,相比Lasso回归对于输入 SNP之间的微小差异具有鲁棒性,且在许多研究中被证实其在预测人的某些复杂表型表现较好[23-24]。Elastic net回归模型、Lasso回归模型、支持向量机回归模型(poly核、RBF核)、Kernel ridge回归模型(poly核、RBF核)、LassoLars回归模型和随机森林回归模型这8个机器学习模型对所有cis-h2区间的基因预测准确性较高。一些非线性模型也被验证在某些时候具有比线性模型高的基因表达预测准确性,如KNN、随机森林回归[25],但在本研究中未发现这些非线性模型总体上具有高于Elastic net回归的预测能力,但是在个别基因上,它们具有较好的预测能力,且在回归模型中使用非线性核RBF和poly相比于线性核能提高模型的预测能力。可能原因是部分基因表达量的SNP效应存在一定的非线性关系。对于cis-h2极低(cis-h2< 0.01)的基因,Lasso回归模型和随机森林回归模型预测能力较强;对于cis-h2中高(cis-h2> 0.20)的基因,Elastic net回归模型和Lasso回归模型预测能力较强。尽管如此,预测效果最好的Elastic net回归模型R2均值小于0.1,预测准确性较低。Fryett等[26]在多个人类数据集上同样使用多种机器学习模型预测基因表达量,将皮尔逊相关系数(r)用于评估预测准确性,发现预测效果最好的BSLMM的r均值为
0.0743 ,预测准确性同样较低。我们认为预测准确性低的原因主要有:虽然我们将基因表达量数据进行校正,但仍包含环境因素等非遗传成分,直接将其与预测得到的GReX计算相关性会造成偏差;同时猪具有复杂的群体和遗传结构,我们使用的机器学习模型并未考虑个体间的亲缘关系。Wainberg等[27]发现预测的基因表达量与实际表达量相关性较低的情况下,TWAS分析的z分数仍然较高,表明预测的表达量在检测关联性上比真实表达量具有优势,能够捕捉到遗传变异、基因表达和性状之间的调控关系。这表明即使预测的基因表达量准确性较低,在TWAS分析中仍具有实质意义。在未来,我们考虑构建混合线性模型,将个体亲缘关系矩阵纳入随机效应,以提升预测准确性。在回归问题中,不同回归模型所适用的最佳特征数会因其内部算法不同而异,在本研究中,我们选取不同cis-SNP数量的基因进行分析,发现一般情况下,cis-SNP数量在一定范围内,随cis-SNP数量即特征数量增加,回归模型拟合效果越好,预测准确性提升,但是SNP数量过少或过多,会使大部分模型在训练时产生欠拟合或过拟合现象,这与Fan等[28]的研究结果相似。所有模型中,线性模型预测准确性受cis-SNP数量影响大于非线性模型。Guyon等[29]的研究中提到在数据是高维的情况下,线性模型往往更依赖于特征选择,意味着线性模型的预测准确性更大程度上受到特征数量的影响,故结果符合预期。
综上所述,相同条件下不同机器学习模型对基因表达量的预测能力存在差异,使用同种机器学习模型的预测准确性受基因cis-h2和cis-SNP数量影响较大。所以根据基因cis-h2和cis-SNP数量选择合适的机器学习模型预测基因表达量,并在未来根据已有数据结构开发新的回归预测模型,能够有效提高预测准确性,进而提高TWAS分析的统计效力。
-
![]()
图 1 不同品种柑橘的果实成熟期果枝的黄龙病症状(A)及砂糖橘枝条的黄龙病四季症状(B)
Figure 1. Typical symptoms of Huanglongbing-affected citrus fruit branches at fruit maturity stage of different cultivars (A) and typical symptoms of Huanglongbing-affected fruit branches of Citrus reticulata cv. Shatangju in different seasons (B)
![]()
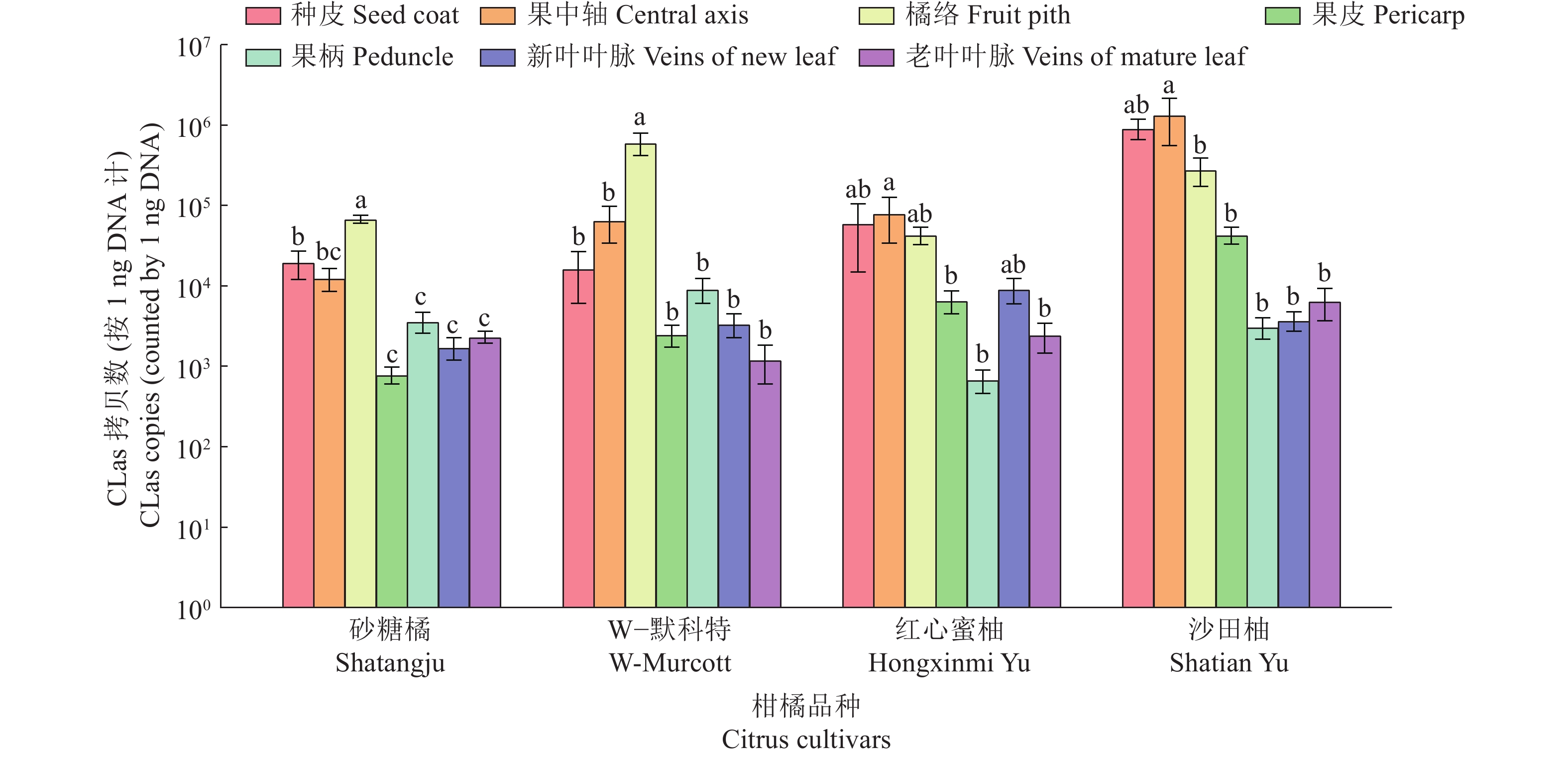
图 2 果实成熟期的不同品种柑橘果枝中的‘Candidatus Liberibacter asiaticus’含量分布
相同品种柱子上的不同字母表示不同部位间差异显著(P<0.05,DMRT法)
Figure 2. Distribution of ‘Candidatus Liberibacter asiaticus’ in Huanglongbing-affected fruit branches of different cultivars at fruit maturity stage
Different lowercase letters on bars of the same cultivar indicate significant differences among different parts (P<0.05,DMRT method)
![]()
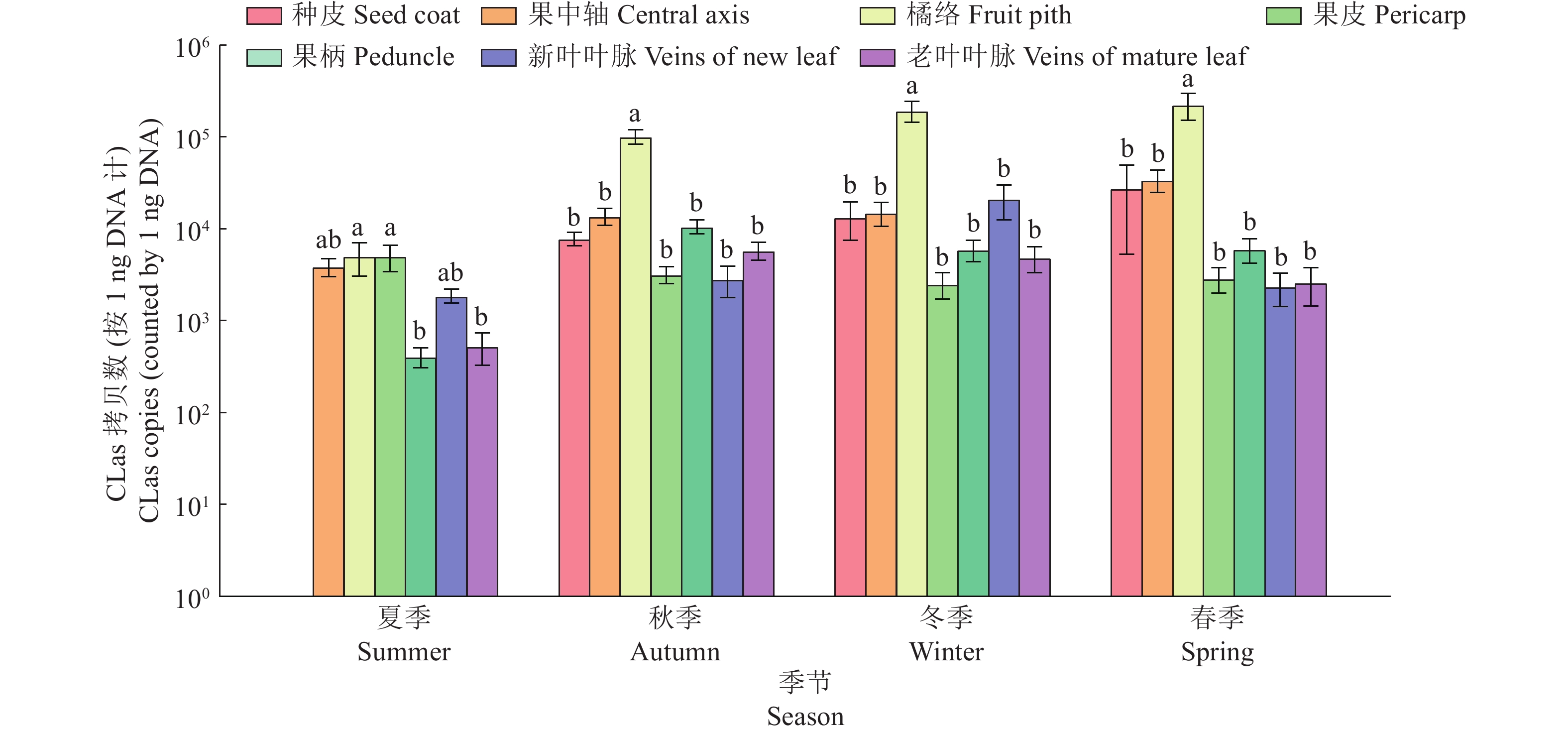
图 3 砂糖橘果枝各部位的‘Candidatus Liberibacter asiaticus’含量随季节变化
相同季节柱子上方的不同小写字母表示不同部位间差异显著(P<0.05,DMRT法)
Figure 3. Quantification of ‘Candidatus Liberibacter asiaticus’ in different parts of Huanglongbing-affected fruit branches of Citrus reticulata cv. Shatangju in different seasons
Different lowercase letters on bars of the same season indicate significant differences among different parts (P< 0.05,DMRT method)
![]()
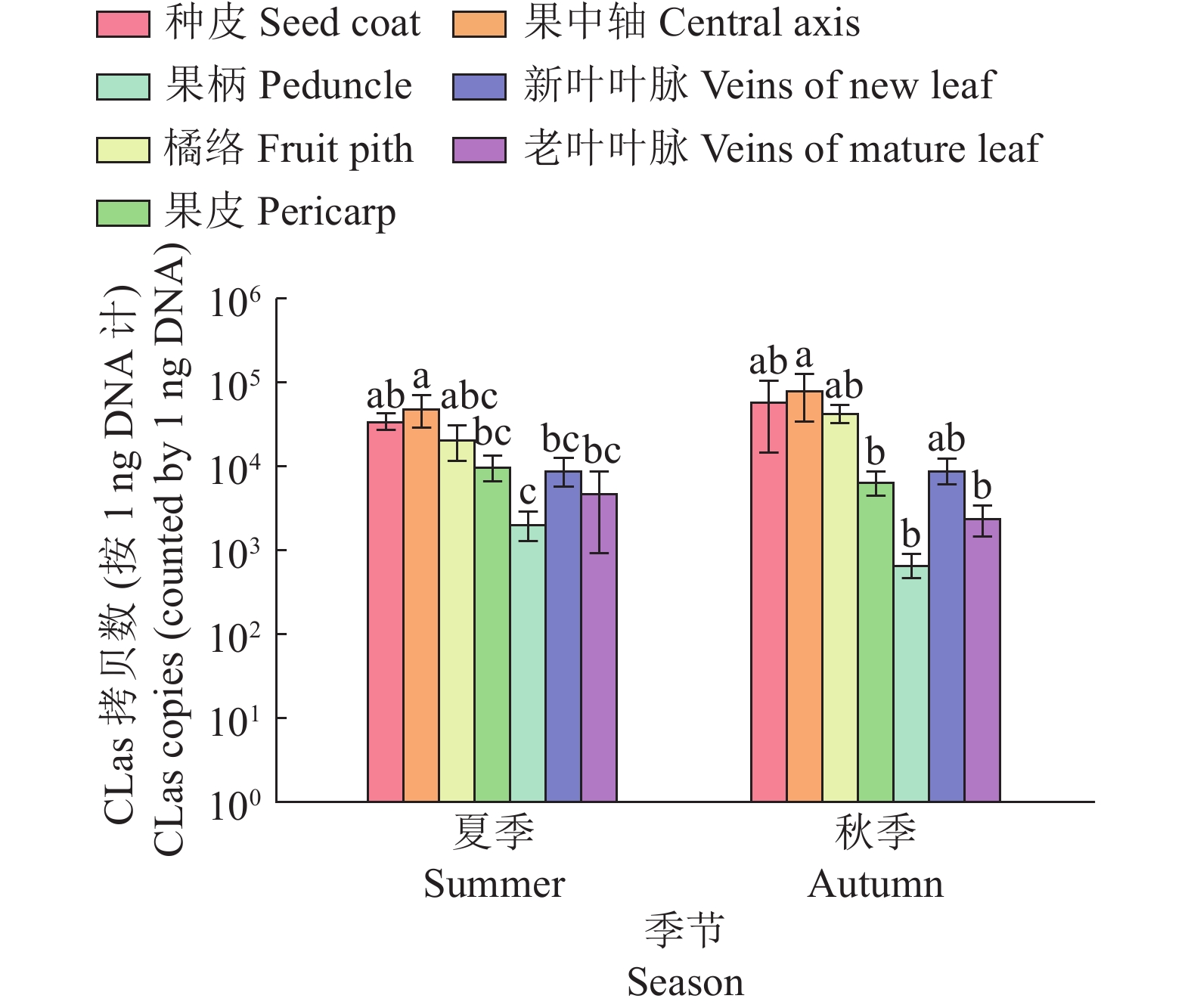
图 4 红心蜜柚果枝各部位的‘Candidatus Liberibacter asiaticus’含量随季节变化
相同季节柱子上的不同小写字母表示不同部位间差异显著(P<0.05,DMRT法)
Figure 4. Quantification of ‘Candidatus Liberibacter asiaticus’ in different parts of Huanglongbing-affected fruit branches of Hongxinmi Yu pumelo in different seasons
Different lowercase letters on bars of the same season indicate significant differences among different parts (P< 0.05,DMRT method)
表 1 本研究的植物材料基本信息
Table 1 Basic information of the plant materials in this study
柑橘品种
Citrus cultivar采集时间
Sampling time采集地点
Sampling location重复
Replications试验1)
Experiment砂糖橘
Citrus reticulata Blanco cv. Shatangju2016-11、 广东博罗
Boluo in Guangdong10 1、2 2017-04/06/10 10 2 红心蜜柚
C. maxima cv. Hongxinmi Yu2017-06/09、 广东博罗
Boluo in Guangdong12 1、2 2020-03 12 1 W−默科特
(C. reticulata ×C. sinensis) cv. W-Murcott2017-06、2021-06 云南瑞丽
Ruili in Yunnan12 1 沙田柚
C. maxima cv. Shatian Yu2016-11、2020-01 广东梅州
Meizhou in Guangdong12 1 1) 1:CLas在不同品种柑橘枝条中的分布情况;2:CLas在不同季节柑橘枝条上的分布规律
1) 1: The distrubution of “Candidatus Liberibacter asiaticus” (CLas) in the fruit branches of different citrus cultivars; 2: The distrubution of CLas in the citrus fruit branches of different seasons 下载: 导出CSV
下载: 导出CSV
-
[1] Center for Agriculture and Bioscience International. Citrus huanglongbing (greening) disease(citrus greening) [EB/OL]. [2021-11-11]. https://www.cabi.org/isc/datasheet/16567.
[2] JAGOUEIX S, BOVÉ J M, GARNIER M. The phloem-limited bacterium of greening disease of citrus is a member of the alpha subdivision of the Proteobacteria[J]. International Journal of Systematic Bacteriology, 1994, 44(3): 379-386. doi: 10.1099/00207713-44-3-379
[3] SAGARAM U S, DEANGELIS K M, TRIVEDI P, et al. Bacterial diversity analysis of Huanglongbing pathogen-infected citrus, using PhyloChip arrays and 16S rRNA gene clone library sequencing[J]. Applied and Environmental Microbiology, 2009, 75(6): 1566-1574. doi: 10.1128/AEM.02404-08
[4] LOPES S A, FRARE G F, BERTOLINI E, et al. Liberibacters associated with citrus Huanglongbing in Brazil: ‘Candidatus Liberibacter asiaticus’ is heat tolerant, ‘ Ca. L. americanus’ is heat sensitive[J]. Plant Disease, 2009, 93(3): 257-262. doi: 10.1094/PDIS-93-3-0257
[5] JOHNSON E G, WU J, BRIGHT D B, et al. Association of ‘Candidatus Liberibacter asiaticus’ root infection, but not phloem plugging with root loss on huanglongbing-affected trees prior to appearance of foliar symptoms[J]. Plant Pathology, 2014, 63(2): 290-298. doi: 10.1111/ppa.12109
[6] WANG N, PIERSON E A, SETUBAL J C, et al. The Candidatus Liberibacter-host interface: Insights into pathogenesis mechanisms and disease control[J]. Annual Review of Phytopathology, 2017, 55(1): 451-482. doi: 10.1146/annurev-phyto-080516-035513
[7] 林孔湘. 柑桔黄梢(黄龙)病研究: Ⅰ: 病情调查[J]. 植物病理学报, 1956, 2(1): 1-11. [8] ACHOR D, WELKER S, BEN-MAHMOUD S, et al. Dynamics of Candidatus Liberibacter asiaticus movement and sieve-pore plugging in citrus sink cells[J]. Plant Physiology, 2020, 182(2): 882-891. doi: 10.1104/pp.19.01391
[9] BOVÉ J M. Huanglongbing or yellow shoot, a disease of Gondwanan origin: Will it destroy citrus worldwide?[J]. Phytoparasitica, 2014, 42(5): 579-583. doi: 10.1007/s12600-014-0415-4
[10] ZHENG Z, CHEN J, DENG X. Historical perspectives, management and current research of citrus HLB in Guangdong province of China, where the disease has been endemic for over a hundred years[J]. Phytopathology, 2018, 108(11): 1224-1236. doi: 10.1094/PHYTO-07-18-0255-IA
[11] TATINENI S, SAGARAM U S, GOWDA S, et al. In planta distribution of ‘Candidatus Liberibacter asiaticus’ as revealed by polymerase chain reaction (PCR) and real-time PCR[J]. Phytopathology, 2008, 98(5): 592-599. doi: 10.1094/PHYTO-98-5-0592
[12] LI W, LEVY L, HARTUNG J S. Quantitative distribution of ‘Candidatus Liberibacter asiaticus’ in citrus plants with citrus huanglongbing[J]. Phytopathology, 2009, 99(2): 139-144. doi: 10.1094/PHYTO-99-2-0139
[13] SAUER A V, ZANUTTO C A, NOCCHI P T R, et al. Seasonal variation in populations of ‘Candidatus Liberibacter asiaticus’ in citrus trees in Paraná State, Brazil[J]. Plant Disease, 2015, 99(8): 1125-1132. doi: 10.1094/PDIS-09-14-0926-RE
[14] LOUZADA E S, VAZQUEZ O E, BRASWELL W E, et al. Distribution of ‘Candidatus Liberibacter asiaticus’ above and below ground in Texas citrus[J]. Phytopathology, 2016, 106(7): 702-709. doi: 10.1094/PHYTO-01-16-0004-R
[15] HU H, ROY A, BRLANSKY R H. Live population dynamics of ‘Candidatus Liberibacter asiaticus’, the bacterial agent associated with citrus huanglongbing, in citrus and non-citrus hosts[J]. Plant Disease, 2014, 98(7): 876-884. doi: 10.1094/PDIS-08-13-0886-RE
[16] 李智鹏, 关巍, 黄洋, 等. 柑橘黄龙病菌在寄主体内含量动态变化研究[J]. 果树学报, 2019, 36(11): 1540-1548. [17] MUNIR S, LI Y, HE P, et al. Seasonal variation and detection frequency of Candidatus Liberibacter asiaticus in Binchuan, Yunnan province China[J]. Physiological and Molecular Plant Pathology, 2019, 106: 137-144. doi: 10.1016/j.pmpp.2019.01.004
[18] 褚丽萍, 郑正, 邓晓玲. 定量分析柑桔黄龙病菌在沙糖桔中的分布[J]. 中国南方果树, 2016, 45(6): 42-43. [19] FANG F, GUO H, ZHAO A, et al. A significantly high abundance of “Candidatus Liberibacter asiaticus” in citrus fruit pith: In planta transcriptome and anatomical analyses[J]. Frontiers in Microbiology, 2021, 12: 1412.
[20] 陈传武, 付慧敏, 邓崇岭, 等. 柑橘和黄皮中黄龙病菌含量动态变化[J]. 南方农业学报, 2015, 46(6): 1024-1028. doi: 10.3969/j.issn.2095-1191.2015.6.1024 [21] 李敏, 鲍敏丽, 黄家权, 等. 柑橘黄龙病菌在寄主根部的消长规律研究[C]//彭友良, 李向东. 中国植物病理学会2017年学术年会论文集. 北京: 中国农业科学技术出版社, 2017: 296. [22] 陈燕玲, 唐瑞, 刘洋, 等. 黄龙病菌在柑橘枝条上的分布和多样性分析[J]. 植物病理学报, 2018, 48(6): 728-737. [23] 张培, 关磊, 刘蕤, 等. 砂糖橘红鼻子果与韧皮部杆菌侵染相关性研究[J]. 热带作物学报, 2011, 32(4): 738-742. doi: 10.3969/j.issn.1000-2561.2011.04.033 [24] IBANEZ F, STELINSKI L L. Temporal dynamics of Candidatus Liberibacter asiaticus titer in mature leaves from Citrus sinensis cv Valencia are associated with vegetative growth[J]. Journal of Economic Entomology, 2019, 113(2): 589-595.
[25] BAO M, ZHENG Z, SUN X, et al. Enhancing PCR capacity to detect ‘Candidatus Liberibacter asiaticus’ utilizing whole genome sequence information[J]. Plant Disease, 2019, 104(2): 527-532.
[26] HARAKAVA R, MARAIS L J, OCHASAN J, et al. Improved sensitiviy in the detection and differentiation of citrus huanglongbing bacteria from South Africa and the Philippines[J]. Cancer Biology & Therapy, 2000, 7(11): 1712-1716.
[27] LOPES S A, LUIZ F Q B F, OLIVEIRA H T, et al. Seasonal variation of ‘Candidatus Liberibacter asiaticus’ titers in new shoots of citrus in distinct environments[J]. Plant Disease, 2017, 101(4): 583-590. doi: 10.1094/PDIS-06-16-0859-RE
[28] ANDRADE M O, PANG Z, ACHOR D S, et al. The flagella of ‘Candidatus Liberibacter asiaticus’ and its movement in planta[J]. Molecular Plant Pathology, 2020, 21(1): 109-123. doi: 10.1111/mpp.12884
[29] 吴柳, 白晓晶, 文庆利, 等. 柑橘黄龙病病原菌CLas在叶圆片嫁接接种的‘锦橙’中早期扩散研究[J]. 园艺学报, 2018, 45(11): 2121-2128. [30] DUAN Y, ZHOU L, HALL D G, et al. Complete genome sequence of citrus huanglongbing bacterium, ‘Candidatus Liberibacter asiaticus’ obtained through metagenomics[J]. Molecular Plant-Microbe Interactions, 2009, 22(8): 1011-1020. doi: 10.1094/MPMI-22-8-1011
[31] 樊晶. 柑橘宿主对黄龙病病原菌侵染的应答机制[D]. 重庆: 重庆大学, 2010.



















































































计量
- 文章访问数: 245
- HTML全文浏览量: 0
- PDF下载量: 393
