Determination of tildipirosin residues in swine tissues by high performance liquid chromatography-tandem mass spectrometry
-
摘要:目的
建立适用于猪组织中泰地罗新残留检测的高效液相色谱−串联质谱法。
方法猪组织(肌肉、肝脏、肾脏和皮脂)样品经0.1 mol·L–1磷酸二氢钾溶液提取,上清液采用HLB固相萃取柱浓缩净化,Phenomenex Luna Omega C18色谱柱梯度洗脱,采用电喷雾正离子扫描和多反应监测(MRM)模式对泰地罗新进行分析,采用基质匹配法对泰地罗新含量进行标准校正。
结果泰地罗新添加剂量在肌肉中25~2 400 ng·g–1、肝脏中25~2 500 ng·g–1、肾脏中25~2 000 ng·g–1和皮脂中25~1 600 ng·g–1的范围内,与特征离子峰面积呈良好的线性关系(r2 ≥ 0.99)。猪组织添加泰地罗新样品中肌肉、肝脏、肾脏和皮脂的检测限均为10 ng·g–1,定量限均为25 ng·g–1。对猪组织在定量限、1/2MRL、MRL、2MRL这4个剂量下进行泰地罗新的添加回收试验,各组织中的批内平均回收率为85.6%~105.0%,批内回收率相对标准偏差2.3%~9.5%,批间回收率相对标准偏差4.7%~7.6%。
结论该方法简单易行,灵敏度高且特异性强,可用于猪组织中泰地罗新残留量的分析测定。
-
关键词:
- 猪组织 /
- 高效液相色谱–串联质谱 /
- 泰地罗新 /
- 残留
Abstract:ObjectiveTo establish a reliable HPLC-MS/MS (High performance liquid chromatography-tandem mass spectrometry) method suitable for determing tildipirosin residues in swine tissues.
MethodSwine tissue samples, including muscle, liver, kidney and skin-fat were extracted with 0.1 mol·L–1 KH2PO4 buffer solution. The supernatant fluids were enriched and purified using HLB solid-phase extraction column, and gradiently eluted by Phenomenex Luna Omega C18 liquid chromatography column. The analytes were then detected using triple-quadrupole mass spectrometry in positive electrospray ionization and multiple reaction monitoring (MRM) mode. The matrix-matched method was used to calibrate tildipirosin content.
ResultTildipirosin contents presented good linear relationships (r2>0.99) with characteristicion peak area in the ranges of 25–2 400 ng·g–1 in muscle, 25–2 500 ng·g–1 in liver, 25–2 000 ng·g–1 in kidney and 25–1 600 ng·g–1 in skin-fat. All the limits of detection and quantitation in muscle, liver, kidney and skin-fat samples added tildipirosin were 10 and 25 ng·g–1, respectively. The recovery experiments of tildipirosin in swine tissues were setted in four dosage levels, including limit of quantitation, 1/2 maximum residue limit (MRL), MRL and 2MRL, and the mean intra-batch recoveries of tildipirosin in all analytes ranged from 85.6% to 105.0%. The relative standard deviations of intra-batch and inter-batch recoveries were in the ranges of 2.3%–9.5% and 4.7%–7.6%, respectively.
ConclusionThe established method is simple, practicable, and with high sensitivity and specificity. It can be applied to determine tildipirosin residues in swine tissues.
-
雷琼黄牛是我国南亚热带的肉牛良种,属华南黄牛体系,主产于广东省湛江市和海南省海口市,分布于邻近县市[1]。雷琼黄牛体貌似印度瘤牛,肩峰隆起、体躯匀称、肌肉结实紧凑,具有极强的抗逆、抗高温高湿、繁殖性能稳定等特征,但由于其体型小、生长速度缓慢等劣势,与我国地方优良的役肉兼用型黄牛品种存在一定的差距[2]。秦川牛、南阳牛、鲁西牛、晋南牛和延边牛5个品种被公认为中国黄牛的代表性品种,其成年公牛体质量分别可达620.9、490.8、512.5、660.0和625.0 kg,而雷琼黄牛仅为354.6 kg[3]。为了更好地发挥雷琼黄牛种资源优势,提高其生长性能与改善其肉品质,育种工作者引进外来优质牛,如西门塔尔牛、利木赞牛、安格斯牛等对雷琼黄牛进行杂交改良,但由于饲养管理水平低,缺乏完善的系谱登记制度,任由外来品种与雷琼黄牛频繁出现杂交紊乱现象,使得杂交牛系谱混乱,导致杂交牛后代杂交优势削减,遗传潜力未得到充分发挥。雷琼黄牛与西门塔尔牛、利木赞牛、安格斯牛杂交F1代生长发育规律与肉品质的研究鲜有报道,本研究以雷琼黄牛及其杂交牛F1代作为研究对象,对各项生长指标(体质量、体高、十字部高、体斜长、胸围)与肉品质理化指标(水分、灰分、蛋白质、脂肪、微量元素、剪切力、眼肌面积、肉色、氨基酸、脂肪酸)进行分析,比较雷琼黄牛及其杂交F1代的生长性状与肉用性能,为肉牛杂交改良提供科学依据。
1. 材料与方法
1.1 试验牛
在广东省湛江市,相同饲养条件下选取发育正常、健康、精神状态良好的西雷杂F1代25头(西门塔尔♂×雷琼黄牛♀)、利雷杂F1代133头(利木赞♂×雷琼黄牛♀)、安雷杂F1代42头(安格斯♂×雷琼黄牛♀)及雷琼黄牛190头,测定其初生、3月龄、6月龄、24月龄的体质量及体尺指标。
随机选取发育正常、健康、精神状态良好24月龄的雷琼黄牛、西雷杂F1代、利雷杂F1代及安雷杂F1代各3头,取其背最长肌为肉样。每头取肉样1 kg装入洁净密封袋中,并放进干冰保温箱中,冷冻保存待测。
1.2 肉牛生产性能测定
1.2.1 生长发育指标测定
测定雷琼黄牛、西雷杂F1代、利雷杂F1代、安雷杂F1代的生长发育性状,包括初生、3月龄、6月龄和24月龄的体质量及体尺指标(体高、十字部高、体斜长、胸围)。
1.2.2 肉品质分析
以雷琼黄牛、西雷杂F1代、利雷杂F1代、安雷杂F1代的背最长肌为样品,测定其理化指标:水分、蛋白质、脂肪、微量元素、氨基酸、脂肪酸和矿物质元素含量,以及剪切力和肉色。
1.2.3 眼肌面积测定
取位于第12和13对胸肋间眼肌,在硫酸纸上描出眼肌横切面轮廓,然后用方格纸描出并获得眼肌面积[4]。
1.2.4 剪切力测定
将眼肌肉块切成23 cm厚的肉块,放置密封袋中,将肉块浸入80 ℃的恒温水浴锅中,加热至肉块中心温度为70 ℃时取出,用直径为1.27 cm的圆柱形取样器避开结缔组织,顺着肌纤维的方向取出。用肌肉嫩度仪测定每个样品的剪切力,每个样品测定8次,选取5个相近的值取其平均值,即为该肉样的剪切力[5]。
1.3 数据分析
应用SPSS 20.0统计分析软件进行数据处理,所有数据结果均取平均值±标准误,采用One-Way ANOVA进行方差分析,采用LSD法进行差异显著性检验。
2. 结果与分析
2.1 雷琼黄牛与不同品种肉牛杂交后代生产性能比较
由表1可知,3组杂交牛F1代的体质量指标相较于雷琼黄牛均有大幅度的提升,其中利雷杂的提升幅度最大。3组杂交牛F1代的初生体质量极显著高于雷琼黄牛(P<0.01),说明3组杂交牛F1代都继承了父本优良的肉用性能,具有较大的初生体质量。利雷杂、西雷杂和安雷杂的初生体质量分别比雷琼黄牛提高了60.55%、51.99%和50.43%,其中利雷杂的提升幅度最大。
表 1 雷琼黄牛与不同品种肉牛杂交F1代生长发育性状1)Table 1. Growth and developmental traits of F1 hybrids of Leiqiong yellow cattle and different breeds of beef cattle牛龄
Age品种
Breed体质量/kg
Body weight体高/cm
Body height十字部高/cm
Hip croop height体斜长/cm
Body length胸围/cm
Chest measurement初生
Birth西雷杂
Simmental×Leiqiong28.24±0.51Aab 65.57±0.78Aab 69.00±1.02Bb 57.71±0.92Aab 70.43±0.61Aab 利雷杂
Limousin×Leiqiong29.83±0.42Aa 66.77±0.34Aa 70.30±0.31Aa 58.93±0.62Aa 70.82±0.40Aa 安雷杂
Angus×Leiqiong27.95±0.32Ab 64.91±0.34Ab 68.00±0.33Bb 56.55±0.28Ab 68.36±0.24Ab 雷琼
Leiqiong18.58±0.35Bc 60.38±0.32Bc 62.71±0.34Cc 53.92±0.30Bc 61.35±0.40Bc 3月龄
3-month-old西雷杂
Simmental×Leiqiong109.50±2.32Aa 86.17±0.95Bb 91.50±1.59Bb 87.00±1.77Aab 104.83±2.90Aab 利雷杂
Limousin×Leiqiong112.93±1.58Aa 87.84±0.61Aa 92.57±0.75Aa 87.16±0.69Aa 106.98±1.10Aa 安雷杂
Angus×Leiqiong93.77±2.51Bb 84.62±0.45Bb 89.00±0.75Bb 84.54±0.33Ab 102.77±0.52Ab 雷琼
Leiqiong67.62±1.16Cc 76.54±0.33Cc 79.59±0.45Cc 76.96±0.34Bc 93.28±0.66Bc 6月龄
6-month-old西雷杂
Simmental×Leiqiong178.14±4.38Bb 95.71±1.17Bb 103.14±1.03Bb 103.00±1.94Bb 127.00±1.05Bb 利雷杂
Limousin×Leiqiong194.33±2.40Aa 98.44±0.46Aa 106.23±0.37Aa 108.28±0.35Aa 130.72±0.48Aa 安雷杂
Angus×Leiqiong160.77±2.32Cc 93.77±0.47Bb 98.77±0.67Cc 97.92±0.71Cc 122.85±1.05Bc 雷琼
Leiqiong140.35±0.76Dd 89.94±0.36Cc 94.74±0.35Dd 100.91±0.67Bb 119.71±0.66Cd 24月龄
24-month-old西雷杂
Simmental×Leiqiong357.05±14.16Bb 122.80±2.50Aab 129.00±2.81Bb 139.00±2.88Aa 175.40±1.72Bb 利雷杂
Limousin×Leiqiong398.37±16.32Aa 123.83±3.00Aa 132.67±3.09Aa 140.17±3.47Aa 184.50±2.28Aa 安雷杂
Angus×Leiqiong331.02±12.05BCb 116.80±0.97ABb 122.80±0.58Bb 125.20±2.62Bb 178.00±1.84Aa 雷琼
Leiqiong289.43±6.56Cc 111.62±0.91Bc 115.77±0.56Cc 120.77±1.80Bb 169.54±1.77Bb 1)相同牛龄同列数据后的不同大、小写字母分别表示差异达到0.01和0.05的显著水平(LSD法)
1)Different capital and lowercase letters in the same column of the same age indicate the difference reaches 0.01 and 0.05 significance levels respectively (LSD method)利雷杂、西雷杂和安雷杂F1代在断乳后不同月龄的体质量显著高于雷琼黄牛(P<0.05),3月龄体质量分别比雷琼黄牛提高了67.01%、61.83%和38.67%,6月龄体质量分别比雷琼黄牛提高了38.46%、26.93%和14.55%,24月龄体质量分别比雷琼黄牛提高了37.64%、23.36%和14.37%。由此可知,3组杂交牛F1代中,利雷杂在初生至24月龄阶段的体质量增加幅度最大。
在初生至24月龄阶段,3组杂交牛F1代和雷琼黄牛的各体尺指标(体高、十字部高、体斜长、胸围)均呈现出不同程度的增长趋势,各体尺指标的增长强度大小顺序总体为利雷杂>西雷杂>安雷杂>雷琼黄牛。由表1可知,利雷杂、西雷杂和安雷杂比同年龄段雷琼黄牛的体高、十字部高、体斜长和胸围都有明显的提升,其中利雷杂的提升幅度是最大的。在24月龄阶段,对于体高、十字部高和体斜长3项体尺指标,利雷杂的测量值均极显著大于安雷杂与雷琼黄牛(P<0.01),西雷杂的测量值显著大于安雷杂与雷琼黄牛(P<0.05);利雷杂与安雷杂的胸围极显著大于雷琼黄牛(P<0.01);而西雷杂和雷琼黄牛的胸围差异不显著;24月龄的利雷杂比雷琼黄牛的体高、十字部高、体斜长、胸围分别提高了10.94%、14.60%、16.06%和8.82%;24月龄的西雷杂比雷琼黄牛的体高、十字部高、体斜长、胸围分别提高了10.02%、11.43%、15.09%和3.46%;24月龄的安雷杂比雷琼黄牛的体高、十字部高、体斜长、胸围分别提高了4.64%、6.07%、3.67%和4.99%。由此可知,3组杂交牛F1代中,利雷杂改良雷琼黄牛体型效果最优。
2.2 雷琼黄牛与不同品种肉牛杂交后代肉品质理化指标分析
2.2.1 肉品质物理指标
消费者主要通过牛肉的嫩度、系水力以及肉质的风味评定肉的好坏,其中嫩度是肉质评定的一个重要指标,高档牛肉剪切力小,易咀嚼[5]。由表2结果可知,剪切力大小顺序为西雷杂>雷琼黄牛>安雷杂>利雷杂,由此可知利雷杂与安雷杂的肉质嫩度优于雷琼黄牛,利雷杂的嫩度最优,而西雷杂的肉质嫩度最差。
表 2 雷琼黄牛与不同品种肉牛杂交F1代的肉品质物理指标1)Table 2. Physical indexes for meat qualities of F1 hybrids of Leiqiong yellow cattle and different breeds of beef cattle品种
Breed剪切力/kg
Shearing force眼肌面积/cm2
Loin eye area肉色 L a b 西雷杂
Simmental×Leiqiong5.80±0.25Aa 73.05±0.63Bb 34.51±0.27Bb 20.09±0.68Aa 13.72±0.75Aa 利雷杂
Limousin×Leiqiong3.71±0.23Cc 60.30±0.43Cc 33.68±0.48Bb 16.89±0.85Bb 11.34±0.67Ab 安雷杂
Angus×Leiqiong4.19±0.24Cc 83.20±0.55Aa 37.05±0.37Aa 16.02±0.93Bb 13.13±0.79Aab 雷琼
Leiqiong5.00±0.20Bb 61.33±0.88Cc 31.85±0.40Cc 12.84±0.58Cc 11.66±0.74Aab 1)同列数据后的不同大、小写字母分别表示差异达到0.01和0.05的显著水平(LSD法)
1)Different capital and lowercase letters in the same column indicate the difference reaches 0.01 and 0.05 significance levels respectively (LSD method)眼肌面积是预测胴体质量的一个重要指标,也是显示瘦肉率比例高低的辅助指标,眼肌面积越大,优质切块比例越大,胴体净肉率越高[6]。由表2可知,本试验中各杂交牛F1代的眼肌面积大小顺序为安雷杂>西雷杂>雷琼黄牛>利雷杂,西雷杂的眼肌面积极显著高于雷琼黄牛(P<0.01),安雷杂的眼肌面积(83.20 cm2)极显著高于其余3组(P<0.01),说明安雷杂和西雷杂的瘦肉率高于雷琼黄牛,安格斯牛在提高雷琼黄牛的瘦肉率方面优势大于西门塔尔牛与利木赞牛,由此可知当培育高档杂交肉牛时,安格斯牛为首选,西门塔尔牛次之。
肉色是牛肉的外观体现,据研究表明,肉色为樱红色能够增加消费者对牛肉的购买欲[7]。L、a、b值分别代表亮度、红度与黄度。由表5可知利雷杂的L值极显著高于雷琼黄牛(P<0.01),安雷杂的L值极显著高于其余3组(P<0.01);利雷杂的a值极显著高于雷琼黄牛(P<0.01);雷琼黄牛的L值与a值都极显著低于3组杂交牛F1代(P<0.01)。由此可知西雷杂、利雷杂与安雷杂3组的肉色优于雷琼黄牛,说明西门塔尔、利木赞和安格斯与雷琼黄牛杂交均可提升雷琼黄牛肉色的红度与亮度,改善雷琼黄牛肉色偏白的情况。
表 5 雷琼黄牛与不同品种肉牛杂交F1代肌肉饱和脂肪酸(SFA)含量1)Table 5. Saturated fatty acid (SFA) contents in muscle of F1 hybrids of Leiqiong yellow cattle and different breeds of beef cattlew/% 品种 Breed 肉豆蔻酸 Myristic acid 棕榈酸 Palmitic acid 硬脂酸 Stearic acid 总量 Total 西雷杂
Simmental×Leiqiong0.84±0.04Dd
24.60±0.36Aa
27.16±0.39Aa
54.24±0.69Aa
利雷杂
Limousin×Leiqiong3.83±0.05Aa
26.88±0.11Aa
17.12±0.05BCc
50.90±0.16Cc
安雷杂
Angus×Leiqiong2.19±0.13Cc
27.78±1.62Aa
24.22±1.68Ab
57.81±2.80Aa
雷琼
Leiqiong2.93±0.27Bb
25.74±1.99Aa
14.34±3.56Cd
49.63±1.92Bb
1)同列数据后的不同大、小写字母分别表示差异达到0.01和0.05的显著水平(LSD法)
1) Different capital and lowercase letters in the same column indicate the difference reaches 0.01 and 0.05 significance levels respectively (LSD method)2.2.2 牛肉营养成分含量
蛋白质与脂肪是牛肉中重要的营养成分[8]。由表3可知,雷琼黄牛的蛋白质含量显著高于利雷杂和西雷杂(P<0.05),与安雷杂差异不显著;雷琼黄牛的脂肪含量显著高于安雷杂和西雷杂(P<0.05)。表明安雷杂F1代保持了安格斯牛肉质中蛋白质含量高、肌内脂肪含量低的特点,通过与安格斯杂交改良降低了原雷琼黄牛的肌内脂肪,提升了雷琼黄牛的瘦肉率。
表 3 雷琼黄牛与不同品种肉牛杂交F1代肌肉营养成分含量1)Table 3. Muscle nutrient contents of F1 hybrids of Leiqiong yellow cattle and different breeds of beef cattle品种
Breedw/% w/(mg·kg−1) 水分
Water灰分
Ash蛋白质
Protein脂肪
LipidNa Fe Mn Ca Zn 西雷杂
Simmental×Leiqiong76.37±0.18Aa 1.06±0.04Aa 23.95±0.41Ac 7.39±0.33Cd 776.67±5.78Aa 18.37±0.78Aa 0.14±0.00Aa 64.87±1.42Aa 48.60±0.32Aa 利雷杂
Limousin×Leiqiong71.30±0.06Ab 1.10±0.00Aa 24.46±0.17Abc 57.66±0.69Aa 608.33±1.76Aa 26.40±2.00Aa 0.11±0.00Aa 43.27±0.74Aa 38.60±0.67Aa 安雷杂
Angus×Leiqiong74.80±0.29Aab 1.13±0.03Aa 25.74±0.25Aab 9.59±0.43Cc 559.67±7.88Aa 16.17±2.29Aa 0.14±0.00Aa 54.03±1.68Aa 34.63±0.15Aa 雷琼
Leiqiong71.60±2.18Ab 1.07±0.03Aa 25.85±0.53Aa 25.36±0.42Bb 455.00±56.20Aa 24.17±3.67Aa 0.11±0.01Aa 21.07±4.78Aa 44.23±2.57Aa 1)同列数据后的不同大、小写字母分别表示差异达到0.01和0.05的显著水平(LSD法)
1) Different capital and lowercase letters in the same column indicate the difference reaches 0.01 and 0.05 significance levels respectively (LSD method)2.2.3 牛肉风味物质含量
肉的风味是评价肉品质好坏的重要指标之一,氨基酸是肉香味的重要前体物质[9]。由表4可知,安雷杂的甘氨酸和丙氨酸含量高于其余3组,表明安格斯牛与雷琼黄牛杂交改良能够提升原雷琼黄牛肉质中风味物质的含量。
表 4 雷琼黄牛与不同品种肉牛杂交F1代肌肉与鲜味有关的氨基酸含量1)Table 4. Amino acid contents in muscle related to umami of F1 hybrids of Leiqiong yellow cattle and different breeds of beef cattlew/% 品种 Breed 谷氨酸 Glu 甘氨酸 Gly 丙氨酸 Ala 天门冬氨酸 Asp 西雷杂
Simmental×Leiqiong13.03±0.14Ab
4.32±0.01Aa
4.31±0.02ABb
6.94±0.04Bc
利雷杂
Limousin×Leiqiong13.88±0.03Aab
3.01±0.06Bb
4.29±0.02Bb
7.70±0.02Ab
安雷杂
Angus×Leiqiong14.41±0.07Aa
4.34±0.01Aa
5.38±0.02Aa
8.12±0.04Aab
雷琼
Leiqiong14.48±0.10Aa
3.99±0.01Bb
4.85±0.02ABab
8.17±0.04Aa
1)同列数据后的不同大、小写字母分别表示差异达到0.01和0.05的显著水平(LSD法)
1)Different capital and lowercase letters in the same column indicate the difference reaches 0.01 and 0.05 significance levels respectively (LSD method)2.2.4 牛肉脂肪酸含量
如表5所示,饱和脂肪酸中,4个品种肉牛背最长肌肉豆蔻酸含量大小顺序为利雷杂>雷琼黄牛>安雷杂>西雷杂,西雷杂的肉豆蔻酸含量(w=0.84%)最小,与其余3组肉牛差异极显著(P<0.01)。4个品种肉牛背最长肌中棕榈酸含量大小顺序为安雷杂>利雷杂>雷琼黄牛>西雷杂,安雷杂棕榈酸含量(w=27.78%)最高,但与其他品种的棕榈酸含量差异不显著。4个品种背最长肌中硬脂酸含量大小顺序为西雷杂>安雷杂>利雷杂>雷琼黄牛,西雷杂硬脂酸含量(w=27.16%)最高,雷琼黄牛硬脂酸含量(w=14.34%)最低,两者差异极显著(P<0.01)。4个品种肉牛饱和脂肪酸总量大小顺序为安雷杂>西雷杂>利雷杂>雷琼黄牛,安雷杂饱和脂肪酸总量(w=57.81%)最高,饱和脂肪酸中棕榈酸和硬脂酸含量较高。
如表6所示,单不饱和脂肪酸中,4个品种肉牛背最长肌肉豆蔻油酸含量大小顺序为利雷杂>雷琼黄牛>西雷杂>安雷杂,利雷杂肉豆蔻油酸含量(w=1.07%)最高,显著高于其余3个品种(P<0.05)。各品种肉牛背最长肌棕榈油酸含量大小顺序为利雷杂>雷琼黄牛>安雷杂>西雷杂,利雷杂棕榈油酸含量(w=5.05%)最高,极显著高于其余3个品种(P<0.01)。油酸是单不饱和脂肪酸中的主要脂肪酸,各品种肉牛间背最长肌中油酸含量大小顺序为利雷杂>雷琼黄牛>安雷杂>西雷杂,利雷杂油酸含量(w=39.11%)最高,极显著高于其余3个品种(P<0.01)。
表 6 雷琼黄牛与不同品种肉牛杂交F1代肌肉单不饱和脂肪酸(MUFA)含量1)Table 6. Monounsaturated fatty acid (MUFA) contents in muscle of F1 hybrids of Leiqiong yellow cattle and different breeds of beef cattlew/% 品种 Breed 肉豆蔻油酸 Myristoleic acid 棕榈油酸 Palmitoleic acid 油酸 Oleic acid 异油酸 Isooleic acid 西雷杂
Simmental×Leiqiong0.55±0.02Ab
1.51±0.05Bb
27.23±1.67Cc
2.95±0.42Aa
利雷杂
Limousin×Leiqiong1.07±0.01Aa
5.05±0.04Aa
39.11±0.14Aa
0.53±0.05Bc
安雷杂
Angus×Leiqiong0.51±0.01Ab
1.71±0.13Bb
33.32±2.37Bb
1.33±0.17Ab
雷琼
Leiqiong0.69±0.18Ab
3.93±0.68Bb
33.50±2.37Bb
1.73±0.58Ab
1)同列数据后的不同大、小写字母分别表示差异达到0.01和0.05的显著水平(LSD法)
1) Different capital and lowercase letters in the same column indicate the difference reaches 0.01 and 0.05 significance levels respectively (LSD method)如表7所示,多不饱和脂肪酸主要有亚油酸和α−亚麻酸,其中亚油酸是主要的多不饱和脂肪酸,各品种肉牛背最长肌中亚油酸大小顺序为西雷杂>雷琼黄牛>安雷杂>利雷杂,西雷杂亚油酸含量(w=7.52%)最高,且极显著高于利雷杂和安雷杂(P<0.01)。各品种肉牛背最长肌α−亚麻酸含量大小顺序为雷琼黄牛>安雷杂>西雷杂>利雷杂,雷琼黄牛α−亚麻酸含量(w=1.94%)最高,且极显著高于其他3个品种(P<0.01)。
表 7 雷琼黄牛与不同品种肉牛杂交F1代肌肉多不饱和脂肪酸(PUFA)含量1)Table 7. Polyunsaturated fatty acid (PUFA) contents in muscle of F1 hybrids of Leiqiong yellow cattle and different breeds of beef cattlew/% 品种 Breed 亚油酸 Linoleic acid α−亚麻酸 α-linolenic acid 花生四烯酸 Arachidonic acid 西雷杂
Simmental×Leiqiong7.52±0.97Aa
0.68±0.06Bb
2.03±0.22Aa
利雷杂
Limousin×Leiqiong1.48±0.02Bb
0.39±0.02Bc
0.12±0.01Bb
安雷杂
Angus×Leiqiong4.20±0.57Bb
0.81±0.14Bb
0.15±0.01Bb
雷琼
Leiqiong6.60±1.80Aa
1.94±0.55Aa
0.49±0.05Bb
1)同列数据后的不同大、小写字母分别表示差异达到0.01和0.05的显著水平(LSD法)
1) Different capital and lowercase letters in the same column indicate the difference reaches 0.01 and 0.05 significance levels respectively (LSD method)2.2.5 牛肉必需氨基酸含量
肉中氨基酸含量和组成是评价肉营养价值的重要指标,也是影响肉品质的重要因素[10]。由表8可知,西雷杂F1代牛肉中必需氨基酸的含量极显著低于雷琼黄牛(P<0.01);而雷琼牛、利雷杂、安雷杂三者的必需氨基酸含量差异不显著。
表 8 雷琼黄牛与不同品种肉牛杂交F1代肌肉必需氨基酸含量1)Table 8. Essential amino acid contents in muscle of F1 hybrids of Leiqiong yellow cattle and different breeds of beef cattlew/% 品种
Breed苏氨酸
Thr缬氨酸
Val亮氨酸
Leu赖氨酸
Lys异亮氨酸
L1e苯丙氨酸
Phe蛋氨酸
Met必需氨基酸
Essential amino acid西雷杂
Simmental×Leiqiong11.34±0.56Ac 12.91±0.31Aa 20.75±0.63Bb 23.64±0.94Bb 11.94±0.51Bb 11.82±0.42Aa 7.48±0.34Aa 88.29±0.53Bb 利雷杂
Limousin×Leiqiong11.70±0.41Ab 12.82±0.41Aa 20.70±0.51Aa 23.40±0.61Aa 12.15±0.45Aa 12.04±0.32Aa 7.20±0.35Aa 88.89±0.35Aa 安雷杂
Angus×Leiqiong11.95±0.22Aa 12.40±0.32Ab 20.74±0.53Aa 23.56±0.73Aa 12.18±0.31Aa 11.95±0.41Aa 7.22±0.41Aa 88.87±0.33Aa 雷琼
Leiqiong11.94±0.31Aa 12.49±0.45Ab 20.92±0.42Aa 23.66±0.53Aa 12.05±0.42Aa 11.39±0.31Aa 7.56±0.32Aa 89.13±0.47Aa 1)同列数据后的不同大、小写字母分别表示差异达到0.01和0.05的显著水平(LSD法)
1) Different capital and lowercase letters in the same column indicate the difference reaches 0.01 and 0.05 significance levels respectively (LSD method)3. 讨论与结论
3.1 西门塔尔牛、安格斯牛、利木赞牛改良雷琼黄牛对其F1代生长性能的影响
雷琼黄牛作为华南地区优良的役用牛,具有耐热性能好、耐粗饲以及抗病能力强等优点。随着农业耕种技术发展以及农业机械化的普及,主要作为役用的雷琼黄牛逐渐转向肉用[11]。由于雷琼黄牛的体型小、生长速度慢以及产肉性能差,导致其经济效益远低于优良的肉用牛品种(如西门塔尔牛、安格斯牛、利木赞牛),因此可以采用杂交改良的方式提高雷琼黄牛的生产性能。
西门塔尔牛、安格斯牛、利木赞牛对雷琼黄牛种牛杂交改良效果明显,其中利雷F1代(利木赞♂×雷琼黄牛♀)在初生体质量、3月龄体质量、6月龄体质量、24月龄体质量和月增质量6个指标上的效果明显。
各杂交F1代的初生质量大小顺序为利雷杂>西雷杂>安雷杂>雷琼黄牛,初生质量分别为29.83、28.24、27.95和18.58 kg,可见,利木赞牛改良本地黄牛F1代的初生质量具有更强的杂交优势。姚守秀等[12]利用利木赞牛、西门塔尔牛、安格斯牛改良北塔山土种牛生长与肉用性能的研究结果表明,利木赞牛、西门塔尔牛、安格斯牛杂交F1代初生质量分别为28.90、26.56、24.95 kg,北塔山土种牛初生质量为20.98 kg[12],与本试验研究结果的趋势相同,本试验中,3组杂交牛F1代都继承了父本初生质量大的特点,其中利雷杂的初生质量大于其余2组杂交牛。
在24月龄阶段,各杂交F1代的体质量大小顺序为利雷杂>西雷杂>安雷杂>雷琼黄牛,其体质量分别为398.37、357.05、331.02和289.43 kg,由试验结果可知,利雷杂的体质量高于西雷杂11.57%,表明利木赞牛改良效果比西门塔尔牛改良效果好。黄力刚等[13]在西门塔尔与利木赞肉牛杂交效果比较分析研究中的结果表明:在断乳期后西门塔尔杂交F1代的体尺指标以及体质量均显著大于利木赞杂交F1代,说明西门塔尔牛的改良效果优于利木赞牛,与本试验的结果不相符,这可能与地方牛的品种、地域环境和饲喂条件有关。
肉牛体尺指标直接反映肉牛的体格大小和体躯结构、发育程度等状况,也间接反映牛组织器官的发育情况及体质量,肉牛体尺指标与家畜的生理机能、生产性能、抗病力以及对外界生活条件的适应能力等密切相关[14]。本试验中,从体尺的改良效果来看,利雷杂的改良效果优于西雷杂与安雷杂,利用利木赞牛改良雷琼黄牛,F1代24月龄的体高、体斜长和胸围分别是123.83、140.17和184.50 cm,从体尺的改良效果上看,利木赞的体尺改良效果更好,利雷杂F1代的体型大于其余3组。
3.2 西门塔尔牛、安格斯牛、利木赞牛改良雷琼黄牛对其F1代牛肉品质的影响
肉的嫩度是肉品质的重要指标,剪切力的大小反映了肉的嫩度,剪切力越小,肉的嫩度越好,高档牛肉的剪切力偏小,嫩度好,易咀嚼[15]。本试验比较了雷琼黄牛与西雷杂、利雷杂和安雷杂F1代背最长肌的剪切力,表现为利雷杂剪切力最小(3.71 kg),西雷杂剪切力最大(5.80 kg),雷琼黄牛和安雷杂剪切力分别为5.00 和4.19 kg。罗欣等[16]在研究中国育肥牛肉品质和感官评定时指出,当牛肉剪切力>5.3 kg时,消费者认为牛肉较韧;当剪切力<4.0 kg时,牛肉较嫩;当剪切力在4.1~5.2 kg时,牛肉嫩度可以接受。对比相关临界值可发现,消费者认为利雷杂F1代的肉质较嫩,安雷杂F1代与雷琼黄牛的嫩度可以接受,西雷杂F1代的肉质嫩度较差。
肉的嫩度受品种、年龄、性别、营养状况以及肌肉部位等因素的影响,其本质是这些因素使肌纤维粗细、质地以及结缔组织质量与数量有着明显的差异,其中肌纤维的粗细及结缔组织的质地是影响肉质嫩度的主要因素[16]。1900年育种学家对利木赞牛进行选育,利木赞牛从兼用型转向专门化肉用品种,并在1924年形成专门化肉牛品种;由于农业机械化以及农耕条件的变化,主要作为役用的雷琼黄牛逐渐转向肉用。刘正柱等[15]在研究中表明,牛的运动量越大,其肌肉的负荷越大,使肌肉的强度提高,肌纤维变粗,导致肌肉剪切力值提高、嫩度降低。专门化肉牛品种利木赞牛(♂)与役用牛雷琼黄牛(♀)杂交改良F1代,其背最长肌嫩度优于母本,符合刘正柱等[15]的研究结果,也与本试验的结果相同。
眼肌面积是决定牛肉等级和产肉量等经济性能的重要指标[17]。崔冰冰等[1]2017年利用安格斯牛与海福特牛改良枣北黄牛,海枣F1代眼肌面积最大(97.53 cm2),安枣F1代眼肌面积次之(95.25 cm2),枣北黄牛眼肌面积最小(81.17 cm2)。本试验中安雷杂F1代的眼肌面积(83.20 cm2)极显著高于其余3组试验牛(P<0.01),但仅比枣北黄牛的眼肌面积提高了2.50%,且远小于海枣F1代与安枣F1代的眼肌面积,说明安雷杂F1代产肉性能尚有大幅提升的空间。
3.3 结论
利木赞牛对雷琼黄牛杂交改良作用明显,尤其在初生体质量、24月龄体质量以及体尺发育方面效果显著。利雷杂F1代肉质嫩度优于其余3组,其中西雷杂F1代肉质嫩度最差;西雷杂F1代的眼肌面积大于雷琼黄牛,安雷杂F1代的眼肌面积大于其余3组;3组杂交牛F1代均可改善雷琼黄牛肉色偏白的情况。利雷杂、安雷杂、雷琼黄牛三者的必需氨基酸含量相比差异不显著,但西雷杂F1代肌肉中必需氨基酸含量极显著低于雷琼黄牛。综合3组杂交牛F1代的生长发育特点与肉品质情况,利雷杂F1代的杂交优势显著,因此利雷杂F1代为最优杂交种。
-
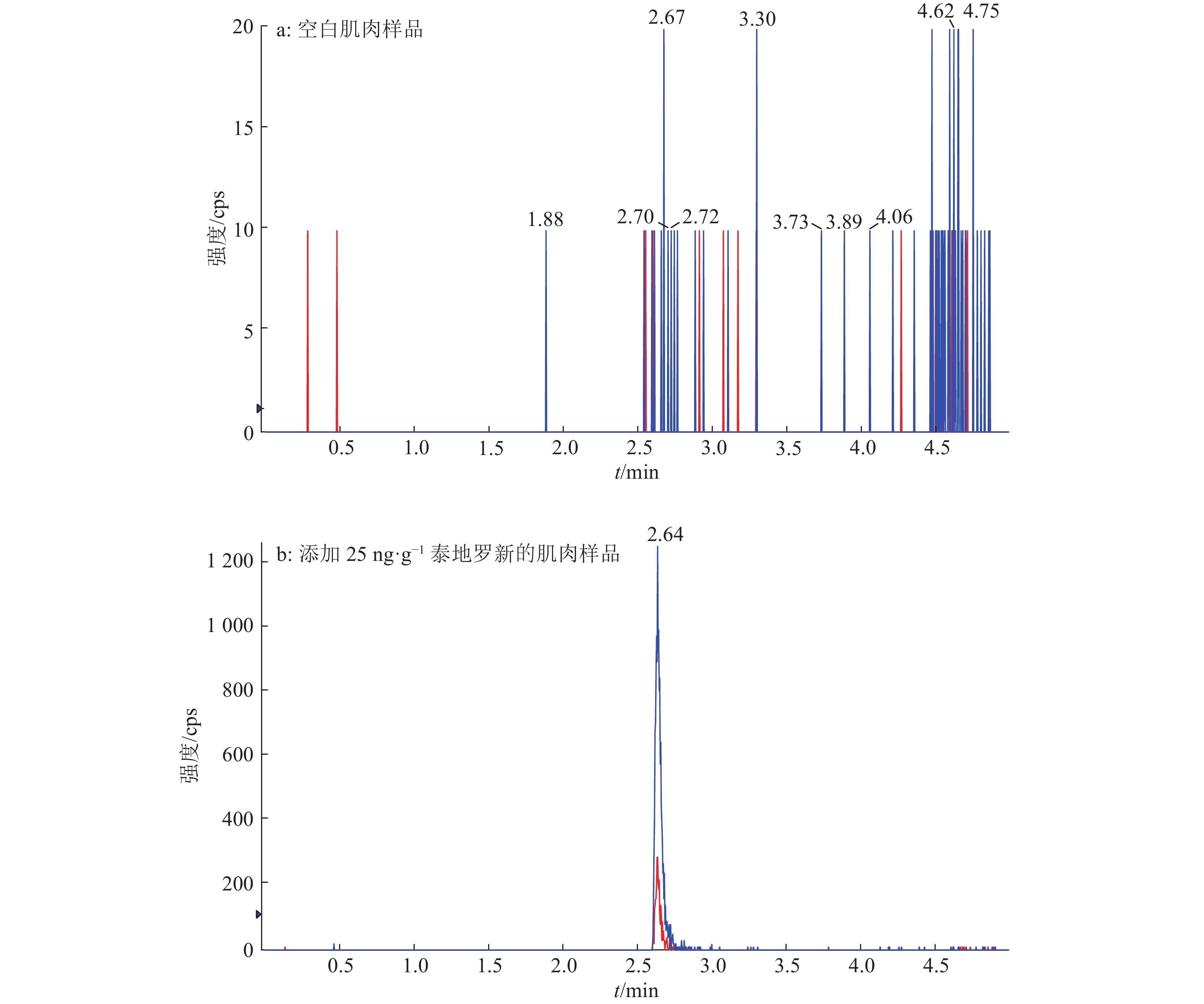
![]()
图 3 空白肌肉样品及添加25 ng·g–1泰地罗新的肌肉样品的多反应检测谱图
Figure 3. Multiple reaction monitoring chromatograms of blank muscle sample and muscle sample added 25 ng·g–1 tildipirosin
表 1 猪组织中泰地罗新检测的基质匹配标准曲线
Table 1 The martrix-matched standard curves of tildipirosin detected in swine tissues
组织样品 w (泰地罗新)1)/
(ng·g–1)标准曲线方程 2) r2 肌肉 25~2 400 y=109x+120 0.995 5 肝脏 25~2 500 y=124x−201 0.998 7 肾脏 25~2 000 y=55.1x+133 0.997 3 皮脂 25~1 600 y=50.8x+214 0.997 5 1)猪组织中泰地罗新的添加剂量范围;2)x:猪组织中泰地罗新的添加剂量,y:泰地罗新特征离子峰面积  下载: 导出CSV
下载: 导出CSV
表 2 猪组织中添加泰地罗新样品回收率及相对标准偏差
Table 2 Recoveries and relative standard deviations of swine tissue samples added tildipirosin
组织
样品w (泰地罗新)添加/
(ng·g–1)批内平均回收率/% 批内回收率相对标准偏差/% 批间平均
回收率/%批间回收率相对
标准偏差/%批次1 批次2 批次3 批次1 批次2 批次3 肌肉 25 101.0 96.6 96.3 6.6 7.3 5.3 97.8 6.4 600 93.3 100.0 97.6 8.5 4.3 8.1 97.1 7.4 1 200 97.5 95.5 97.9 9.5 5.6 6.6 97.0 7.1 2 400 99.0 98.5 99.6 4.7 7.9 6.3 99.1 6.1 肝脏 25 99.1 96.8 101.0 6.7 8.0 7.1 99.1 7.1 2 500 99.9 99.1 101.0 6.4 6.8 3.1 100.0 5.4 5 000 96.0 101.0 93.3 5.8 2.3 4.6 96.7 5.3 10 000 96.7 100.0 93.9 5.4 2.6 6.2 96.9 5.3 肾脏 25 101.0 98.4 101.0 4.3 8.9 6.1 100.0 6.4 5 000 92.3 96.1 97.2 6.0 4.9 2.9 95.2 5.0 10 000 85.6 91.5 93.8 2.5 4.5 4.5 90.3 5.5 20 000 93.3 90.0 96.1 4.9 3.3 3.7 93.1 4.7 皮脂 25 100.0 105.0 102.0 6.3 4.2 4.8 102.0 5.2 400 94.6 102.0 90.7 3.2 4.1 4.6 95.8 6.3 800 92.3 94.4 91.1 8.2 5.9 6.5 92.6 6.7 1 600 92.4 94.7 91.5 9.0 5.6 8.8 92.9 7.6
下载: 导出CSV
-
[1] VANNUFFEL P, COCITO C. Mechanism of action of streptogramins and macrolides[J]. Drugs, 1996, 51(Suppl 1): 20-30.
[2] ROSE M, MENGE M, BOHLAND C, et al. Pharmacokinetics of tildipirosin in porcine plasma, lung tissue, and bronchial fluid and effects of test conditions on in vitro activity against reference strains and field isolates of Actinobacillus pleuropneumoniae[J]. J Vet Pharmacol Ther, 2013, 36(2): 140-153. doi: 10.1111/jvp.2013.36.issue-2
[3] TORRES F, SANTAMARIA R, JIMENEZ M, et al. Pharmacokinetics of tildipirosin in pig tonsils[J]. J Vet Pharmacol Ther, 2016, 39(2): 199-201. doi: 10.1111/jvp.2016.39.issue-2
[4] MENGE M, ROSE M, BOHLAND C, et al. Pharmacokinetics of tildipirosin in bovine plasma, lung tissue, and bronchial fluid (from live, nonanesthetized cattle)[J]. J Vet Pharmacol Ther, 2012, 35(6): 550-559. doi: 10.1111/jvp.2012.35.issue-6
[5] LEAL C, CODONY R, COMPAÑÓ R, et al. Determination of macrolide antibiotics by liquid chromatography[J]. J Chromatogr A, 2001, 910(2): 285-290. doi: 10.1016/S0021-9673(00)01231-0
[6] GARCÍA-MAYOR M A, GARCINUÑO R M, FERNÁNDEZ-HERNANDO P, et al. Liquid chromatography-UV diode-array detection method for multi-residue determination of macrolide antibiotics in sheep's milk[J]. J Chromatogr A, 2006, 1122(1/2): 76-83.
[7] HORIE M, SAITO K, ISHII R, et al. Simultaneous determination of five macrolide antibiotics in meat by high-performance liquid chromatography[J]. J Chromatogr A, 1998, 812(1/2): 295-302.
[8] TORANO J S, GUCHELAAR H J. Quantitative determination of the macrolide antibiotics erythromycin, roxithromycin, azithromycin and clarithromycin in human serum by high-performance liquid chromatography using pre-column derivation with 9-fluorenylmethyloxycarbonyl chloride and fluorescence detection[J]. J Chromatogr B, 1998, 720(1/2): 89-97.
[9] De La HUEBRA M J G, VINCENT U, VON HOLST C. Sample preparation strategy for the simultaneous determination of macrolide antibiotics in animal feedingstuffs by liquid chromatography with electrochemical detection (HPLC-ECD)[J]. J Pharm Biomed Anal, 2007, 43(5): 1628-1637. doi: 10.1016/j.jpba.2006.12.008
[10] SONG X, ZHOU T, LI J, et al. Determination of macrolide antibiotics residues in pork using molecularly imprinted dispersive solid-phase extraction coupled with LC-MS/MS[J]. J Sep Sci, 2018, 41(5): 1138-1148. doi: 10.1002/jssc.v41.5
[11] SONG X, ZHOU T, LIU Q, et al. Molecularly imprinted solid-phase extraction for the determination of ten macrolide drugs residues in animal muscles by liquid chromatography-tandem mass spectrometry[J]. Food Chem, 2016, 208: 169-176. doi: 10.1016/j.foodchem.2016.03.070
[12] LI X, WEN K, CHEN Y, et al. Multiplex immunogold chromatographic assay for simultaneous determination of macrolide antibiotics in raw milk[J]. Food Anal Methods, 2015, 8(9): 2368-2375. doi: 10.1007/s12161-015-0130-x
[13] TAKATSUKI K, USHIZAWA I, SHOJI T. Gas chromatographic-mass spectrometric determination of macrolide antibiotics in beef and pork using single ion monitoring[J]. J Chromatogr A, 1987, 391(1): 207-217.
[14] HELLER D N, NOCHETTO C B. Development of multiclass methods for drug residues in eggs: Silica SPE cleanup and LC-MS/MS analysis of ionophore and macrolide residues[J]. J Agric Food Chem, 2004, 52(23): 6848-6856. doi: 10.1021/jf040185j
[15] WANG J, LEUNG D, BUTTERWORTH F. Determination of five macrolide antibiotic residues in eggs using liquid chromatography/electrospray ionization tandem mass spectrometry[J]. J Agric Food Chem, 2005, 53(6): 1857-1865. doi: 10.1021/jf048414p
[16] WANG J, LEUNG D, LENZ S P. Determination of five macrolide antibiotic residues in raw milk using liquid chromatography-electrospray ionization tandem mass spectrometry[J]. J Agric Food Chem, 2006, 54(8): 2873-2880. doi: 10.1021/jf060068j
[17] 王敏, 林维宣, 郭德华, 等. 高效液相色谱−串联质谱法同时检测动物性食品中多种大环内酯类药物[J]. 分析测试学报, 2007, 26(5): 675-678. doi: 10.3969/j.issn.1004-4957.2007.05.018 [18] 岳振峰, 陈小霞, 谢丽琪, 等. 高效液相色谱串联质谱法测定动物组织中林可酰胺类和大环内酯类抗生素残留[J]. 分析化学, 2007, 35(9): 1290-1294. doi: 10.3321/j.issn:0253-3820.2007.09.010 [19] TAKEGAMI H, HORIE M, NAKAZAWA H. Determination of macrolide antibiotics in milk by high-performance liquid chromatography/mass spectrometry[J]. Bunseki Kagaku, 2006, 55(9): 651-659. doi: 10.2116/bunsekikagaku.55.651
[20] CHERLET M, De BAERE S, CROUBELS S, et al. Quantitation of tylosin in swine tissues by liquid chromatography combined with electrospray ionization mass spectrometry[J]. Anal Chim Acta, 2002, 473(1/2): 167-175.
[21] HORIE M, TAKEGAMI H, TOYA K, et al. Determination of spiramycin and tilmicosin in meat and fish by LC/MS[J]. Shokuhin Eiseigaku Zasshi, 2003, 44(3): 150-154. doi: 10.3358/shokueishi.44.150
[22] GRANELLI K, BRANZELL C. Rapid multi-residue screening of antibiotics in muscle and kidney by liquid chromatography-electrospray ionization-tandem mass spectrometry[J]. Anal Chim Acta, 2007, 586(1/2): 289-295.
[23] WU J, QIAN Y, ZHANG C, et al. Application of graphene-based solid-phase extraction coupled with ultra high-performance liquid chromatography-tandem mass spectrometry for determination of macrolides in fish tissues[J]. Food Anal Methods, 2013, 6(5): 1448-1457. doi: 10.1007/s12161-013-9563-2
[24] TAO Y, YU G, CHEN D, et al. Determination of 17 macrolide antibiotics and avermectins residues in meat with accelerated solvent extraction by liquid chromatography-tandem mass spectrometry[J]. J Chromatogr B Analyt Technol Biomed Life Sci, 2012, 897(1): 64-71.
[25] CHEN K Y, YANG T C, CHANG S Y. Determination of macrolide antibiotics using dispersive liquid-liquid microextraction followed by surface-assisted laser desorption/ionization mass spectrometry[J]. J Am Soc Mass Spectrom, 2012, 23(6): 1157-1160. doi: 10.1007/s13361-012-0371-5
[26] DICKSON L C. Performance characterization of a quantitative liquid chromatography-tandem mass spectrometric method for 12 macrolide and lincosamide antibiotics in salmon, shrimp and tilapia[J]. J Chromatogr B Analyt Technol Biomed Life Sci, 2014, 967: 203-210. doi: 10.1016/j.jchromb.2014.07.031
[27] YANG S W, CHA J, CARLSON K. Quantitative determination of trace concentrations of tetracycline and sulfonamide antibiotics in surface water using solid-phase extraction and liquid chromatography/ion trap tandem mass spectrometry[J]. Rapid Commun Mass Spectrom, 2004, 18(18): 2131-2145. doi: 10.1002/(ISSN)1097-0231
[28] WANG J. Determination of five macrolide antibiotic residues in honey by LC-ESI-MS and LC-ESI-MS/MS[J]. J Agric Food Chem, 2004, 52(2): 171-181. doi: 10.1021/jf034823u
[29] McCLURE E L, WONG C S. Solid phase microextraction of macrolide, trimethoprim, and sulfonamide antibiotics in wastewaters[J]. J Chromatogr A, 2007, 1169(1/2): 53-62.
-
期刊类型引用(10)
1. 邓永涛,尹苗,王聪,陈希文. 猪舍环境影响因子预测模型的研究进展. 家畜生态学报. 2025(02): 122-128 .  百度学术
百度学术
2. 刘艳昌,吴延昌,郭颖轩,李冬阳,张志霞,左现刚,李国厚. 生猪生长环境舒适度实时监测系统设计与实现. 家畜生态学报. 2024(06): 76-81+96 . 百度学术
3. 刘丙杰,侯慕馨,冀海燕. 基于支持向量机集成的船舶舱室温湿度预测. 海军工程大学学报. 2024(03): 21-25+32 . 百度学术
4. 朱佳明,孙彬,蒲施桦,潘学民,徐顺来,胡彬,齐仁立. 基于多环境因素分析的猪舍温湿度预测模型. 华南农业大学学报. 2024(05): 709-721 . 本站查看
5. 杨晓平,李雪寒,聂鹏程. 设施生猪环境调控现状研究和未来展望. 中国农业信息. 2024(02): 83-96 . 百度学术
6. 尤德安,邬锡权,陆畅,董钊杰,曾志雄,吕恩利. 数字孪生技术在现代化猪场生产应用的研究进展. 现代农业装备. 2023(04): 2-9+70 . 百度学术
7. 翟子鹤,陈小文,高莉平,张天柱. 基于梯度提升算法的温室黄瓜株高生长模拟. 中国农业大学学报. 2022(05): 134-145 . 百度学术
8. 杨东轩,吴叶兰,张刚刚,刘硕. 基于边缘计算与深度学习的禽舍监测系统设计. 江苏农业科学. 2022(09): 219-225 . 百度学术
9. 陈谦,杨涵,王宝刚,李文生,钱建平. 基于GRU神经网络模型的冷链运输温度时序预测. 农业大数据学报. 2022(01): 82-88 . 百度学术
10. 刘炫文,刘仁鑫. 猪舍小气候环境模拟预测技术研究综述. 江西农业学报. 2022(08): 122-126 . 百度学术
其他类型引用(7)