
Over-expression of rabies virus G protein and its inhibitory effect on the virus in neuroblastoma cells
-
摘要:目的
探究G蛋白在狂犬病病毒(Rabies virus,RABV)复制中的作用,以揭示携带双G基因的重组RABV的Hep-dG与亲代毒株rHep-Flury在神经母细胞瘤(NA)细胞中滴度差异的原因,为RABV致病机制的研究奠定基础。
方法通过病毒吸附、入侵、荧光定量PCR、Western-blot以及中和抗体阻断等试验,检测G蛋白过表达对IFN-β以及相关因子转录的影响。
结果Hep-dG感染能显著上调NA细胞中IFN-β mRNA的表达,激活了下游因子STAT1的表达与磷酸化,在较低的感染复数(MOI=0.01)下,Hep-dG感染后24 h即可显著促进IFN-β 基因的表达,36 h达到最高水平(P<0.001)。该病毒进入细胞后,产生了更多的病毒Leader RNA和RIG-I mRNA,且与IFN-β mRNA的表达高度一致。抗体阻断IFN-β后,Hep-dG在NA细胞中的病毒滴度显著上升(P<0.01),约为阻断前的7.9倍,且与亲代毒株rHep-Flury无显著差异。与阴性对照比较,5 μg的pH-G质粒转染能刺激IFN-β的转录(P<0.05),表明真核表达RABV G蛋白能在一定程度上刺激IFN-β的转录。
结论本研究初步揭示了G蛋白激活先天性免疫应答的原因和作用。RABV G蛋白的过表达,通过促进病毒Leader RNA的转录,激活了RIG-I介导的IFN-β通路,进而抑制了Hep-dG在NA细胞的繁殖。G蛋白的过表达也对干扰素通路起到一定的作用。
Abstract:ObjectiveTo explore the role of G protein in rabies virus (RABV) replication, reveal the reason for the difference of virus titer in neuroblastoma (NA) cells between the recombinant RABV Hep-dG with dual copy of G gene and the parental strain rHep-Flury, and lay a foundation for the study of RABV pathogenesis.
MethodThe effects of G protein over-expression on transcriptions ofIFN-β and related factors were examined by the virus binding assay, virus entry assay, fluorescence quantitative PCR, Western-blot and neutralizing antibody blocking assay.
ResultHep-dG infection significantly increased the expression of IFN-β mRNA and activated the expression of the downstream factor STAT1 in NA cells. Under the low multiplicity of infection (MOI=0.01), the expression of IFN-β gene significantly increased at 24 h after Hep-dG infection and reached the highest level at 36 h. After the virus entered the cells, there were more viral Leader RNA and RIG-I mRNA, which were highly consistent with the expression of IFN-β mRNA. The block of IFN-β expression by neutralizing antibody in NA cells significantly increased the virus titer of Hep-dG in cell culture supernatant(P<0.01), which was 7.9 times before blocking. Meanwhile the virus titer of Hep-dG had no significant difference with the parental strain rHep-Flury. Compared with the negative control, transfection of 5 μg pH-G plasmid could stimulate the transcription ofIFN-β(P<0.05), which showed that eukaryotic expression of RABV G protein could stimulateIFN-β transcription to a certain extent.
ConclusionThis study preliminarily reveals the cause and role of G protein in activating innate immune response. Over-expression of RABV G protein activates the RIG-I-mediated IFN-β pathway by promoting transcription of the viral Leader RNA, which in turn inhibits Hep-dG replication in NA cells and finally results in the lower virus titer in NA cells.
-
Keywords:
- rabies virus /
- glycoprotein /
- IFN-β pathway /
- interferon-β /
- virus titer
-
在基因工程相关研究中,对淀粉合成相关酶及基因方面的研究已经取得了一定的进展[1],但启动子作为基因表达的重要调控区域,在淀粉相关研究中报道相对较少。选择合适的启动子是有目的地表达外源基因面临的重要科学问题,也是培育安全、高效转基因作物的首要问题[2]。淀粉是玉米Zea mays中的主要物质,玉米中淀粉含量的高低直接影响玉米的产量[3],直链淀粉与支链淀粉的比例影响淀粉的品质[4],玉米淀粉是一种优良并且可靠的淀粉来源,全世界80%淀粉来源于玉米[5],玉米淀粉在化工、医药、纺织、造纸和建筑等领域得到广泛的应用,淀粉的需求量在日益增加,因此提高玉米淀粉含量和改良玉米淀粉品质已成为重要议题,对于玉米淀粉的研究具有显著的社会效益和经济效益[6-7]。
淀粉是在胚乳造粉质体中经一系列酶的配合作用合成的,其中淀粉合成酶在其合成中起到决定性作用,可溶型淀粉合成酶主要包括SSⅠ、SSⅡ、SSⅢ[8-10],其中SSⅠ和SSⅢ的表达已相继被证明具有胚乳特异性。玉米淀粉合成酶SSⅡa是淀粉合成酶中的关键酶,能够参与玉米支链淀粉的合成[11],对其启动子的研究有助于从调控水平上改善及改变SSⅡa活性及作用,进而实现增产及改良目的,完善淀粉代谢网络调控的研究。
本研究克隆并分析SSⅡa启动子,构建5个缺失体植物表达载体,采用三亲杂交法将目标质粒转入农杆菌Agrobacterium tumefacies,用蘸花法将农杆菌转入拟南芥Arabidopsis thaliana,使用除草剂筛选获得阳性植株。PCR检测鉴定拟南芥阳性植株,采用GUS染色和gus基因定量分析启动子的作用部位和活性,以期通过基因工程手段提高玉米淀粉含量,改良淀粉品质提供候选特异性启动子,并为玉米SSⅡa启动子功能研究提供理论依据。
1. 材料与方法
1.1 材料与试剂
玉米B73种子由吉林省农业科学院农业生物技术研究所提供,盆栽砂培,取三叶期的叶片提取基因组DNA,-20 ℃保存备用。植物表达载体pCAMBIA3301由吉林农业大学生命科学学院生物化学与分子生物学实验室保存。
基因组DNA提取试剂盒(Bio Teke)、限制性内切酶EcoRⅠ、BglⅡ和LA Taq均购自TaKaRa生物有限公司,质粒提取试剂盒及DNA凝胶回收试剂盒为AxyGEN公司产品。
克隆SSⅡa基因所用上游引物SSⅡaF:5′-TGTCAGACTGGTTAGTGGAGC-3′;下游引物SSⅡaR:5′-AGAAGGTGGAGGAAGAGGACG-3′。
构建不同长度启动子表达载体的上游引物如下:
SSⅡaF1:5′-CGGAATTCCCTTGACTGGCATCCTT-CCTA-3′,
SSⅡaF2:5′-CGGAATTCTAGAAAGATGTCCCAC-AGAGA-3′,
SSⅡaF3:5′-CGGAATTCTAGCCTATGCTTACCTT-TCAG-3′,
SSⅡaF4:5′-CGGAATTCACGCCATTTTCCATCGT-GCCA-3′,
SSⅡaF5:5′-CGGAATTCCTCGCTGGGCTGCCGTA-GGTA-3′,
共同的下游引物为SSⅡaR:5′-GAAGATCTGG-CGGCGGGATCGATCG-3′。
下划线分别为EcoR I(GAATTC)和BglⅡ(AGATCT)的酶切位点序列。
gus基因检测所用上游引物GUS-F:5′-TTCCTGATTAACCACAAACC-3′;下游引物GUS-R:CGGTTCGTTGGCAATACTCC。
gus基因的定量分析所用β-Actin内参基因上游引物Actin-F:5′-TGCCAATCTACGAGGGTTTC-3′;下游引物Actin-R:5′-GCTCTGCTGTTGTGGTGAAC-3′;目的gus基因上游引物qGUS-F:5′-CTCACACCG-ATACCATCAGC-3′; 下游引物qGUS-R:5′-TACCTT-CTCTGCCGTTTCCA-3′。
1.2 SSⅡa启动子的克隆及序列分析
利用试剂盒提取玉米B73三叶期叶片基因组DNA,根据玉米数据库(http://www.maizegdb.org.ssr/)公布信息,BLAST找到SSⅡa基因5′侧翼序列,利用Primer 5.0软件设计克隆引物SSⅡaF和SSⅡaR,采用常规PCR方法扩增SSⅡa基因5′侧翼序列,PCR条件为:95 ℃ 5 min; 94 ℃ 30 s,69.7 ℃ 40 s,72 ℃ 150 s,35个循环;72 ℃,15 min。扩增产物连接到pMD-18T载体上,转化到大肠埃希菌DH5α,挑取单菌落经PCR鉴定后送至上海生工生物工程公司测序,测序结果与已知序列比对后,利用PlantCare(http://bioinformatics.psb.ugent.be/webtools/plantcare/html/)启动子在线分析软件分析及预测启动子顺式作用元件等功能元件信息[12]。
1.3 SSⅡa启动子植物表达载体的构建
克隆的SSⅡa启动子经序列分析后,依据功能元件所在位置,设计5对引物SSⅡaF1和SSⅡaR、SSⅡaF2和SSⅡaR、SSⅡaF3和SSⅡaR、SSⅡaF4和SSⅡaR、SSⅡaF5和SSⅡaR,并在上下游引物两侧分别加上EcoRⅠ和BglⅡ酶切位点,扩增SSⅡa启动子序列的长度分别为1 407、867、633、483和365 bp,用于构建5个不同长度SSⅡa启动子缺失的植物表达载体[13]。扩增5种长度启动子序列的退火温度分别为56.5、56.5、56.5、62.9和62.9 ℃。将PCR产物与植物表达载体pCAMBIA3301分别双酶切后,16 ℃过夜连接,构建5种重组载体(命名为:P1、P2、P3、P4和P5),分别转化大肠埃希菌DH5α,挑取经PCR及酶切验证后的阳性克隆菌液,送至上海生工生物工程公司测序。
1.4 拟南芥的遗传转化
哥伦比亚型拟南芥,农杆菌EHA105和含HELP质粒菌株均由吉林农业大学生命科学学院生物化学与分子生物学实验室提供,德国泥炭土、蛭石、MS盐、SilwetL-77、6-苄氨基嘌呤(6-BA)、B5维生素、蔗糖、利福平(Rif)抗生素、Kan抗生素等试剂,以及LB培养基和YEP培养基均购置于上海生工生物工程公司。
三亲杂交法将重组植物表达载体导入农杆菌,植物材料培养于人工气候室,拟南芥长至开花期采用农杆菌介导的花序侵染法侵染拟南芥,收获种子,种植,用除草剂筛选阳性植株,作为功能验证试验材料[13]。
1.5 SSⅡa启动子在拟南芥中的功能分析
1.5.1 gus基因的PCR检测
提取含5种不同类型启动子的转基因拟南芥阳性植株(以非转基因拟南芥为对照)的叶片基因组DNA,以其为模板,植物表达载体pCAMBIA3301上的gus基因为目标基因设计的引物(GUS-F, GUS-R)进行PCR扩增,序列长度为402 bp,经10 g·L-1琼脂糖凝胶电泳检测。
1.5.2 转基因植株的GUS组织化学分析
分别取转P1、P2、P3、P4和P5载体的阳性植株和野生型拟南芥,成熟期后分别取叶片和果荚进行GUS染色,参照Jefferson方法[14](略有改动),具体步骤包括:
1) 清洗:使用无菌水清洗样品,去除样品表面杂物;
2)染色:将材料放置培养皿中,使样品完全浸入到GUS染色液中,置于37 ℃培养箱中过夜;
3) 脱色:将染色材料置于体积分数为80%的乙醇溶液中脱色,期间视乙醇溶液颜色及时更换乙醇溶液,直至拟南芥材料完全脱色(绿色褪去,呈现白色)。脱色后的材料拍照观察,记录。
1.5.3 gus基因的定量分析
提取上述不同试验材料成熟期叶片和种子的RNA,反转录成cDNA为模板,以β-Actin基因为内参,实时荧光定量PCR法测定gus基因的表达,3次重复,采用2-ΔΔCt法进行分析,计算gus基因的相对表达量[15]。
2. 结果与分析
2.1 SSⅡa启动子克隆及分析结果
用玉米B73基因组DNA为模板,SSⅡaF, SSⅡaR为引物进行克隆,得到2 526 bp序列(见图 1A),与pMD18T连接,转化后摇菌,菌液PCR验证结果如图 1B所示,将验证好的阳性克隆测序表明,获得的2 526 bp中有1个碱基突变,与参考序列比对一致性为99.96%,经PlantCare软件分析表明此碱基的差异并未在启动子功能元件处,不影响启动子的功能。
![]() 图 1 SSⅡa启动子克隆和PCR验证的电泳结果M:DL2000 DNA Marker;1~3:SSⅡa启动子。Figure 1. Electrophoresis results of SSⅡa promoter cloning and PCR amplification
图 1 SSⅡa启动子克隆和PCR验证的电泳结果M:DL2000 DNA Marker;1~3:SSⅡa启动子。Figure 1. Electrophoresis results of SSⅡa promoter cloning and PCR amplification核酸测序结果及功能元件分析结果如图 2所示,经PlantCare软件分析发现,SSⅡa序列含有众多启动子必需的顺式作用元件,具体元件信息如表 1所示,其中包括TATA-box,CAAT-box等元件。该序列存在9个TATA-box,离SSⅡa基因ATG最近的为处于504 bp处的TATA-box(图 2红色方框中显示),除此之外,分析还发现了众多重要的功能元件和结合位点(表 1),例如1)MBS:MYB结合位点,能够响应水分、盐、低温等逆境胁迫;2)Skn-1-motif和GCN4-motif:胚乳表达所需的顺式作用元件;3)motif IIb:参与脱落酸响应的功能元件;4)O2-site:参与玉米醇溶蛋白代谢调控的功能元件。这些功能元件的存在,说明玉米SSⅡa基因能够受到多种生物及非生物胁迫的诱导及影响,而且拥有胚乳特异性表达的功能元件。该启动子相关功能元件的研究,对于玉米SSⅡa基因的表达调控途径的探究及开发胚乳特异型启动子具有重要意义。
![]() 图 2 玉米SSⅡa启动子的序列分析附有黄色底纹的序列为相关顺式作用元件,由上至下红色方框内依次为克隆突变碱基、胚乳特异性表达功能元件、参与抗旱诱导的MYB结合位点、TATA-box、SSⅡa基因起始密码子ATG。Figure 2. Sequence analysis of maize SSⅡa promoter表 1 SSⅡa启动子区顺式作用元件Table 1. Cis-acting elements of SSⅡa promoter
图 2 玉米SSⅡa启动子的序列分析附有黄色底纹的序列为相关顺式作用元件,由上至下红色方框内依次为克隆突变碱基、胚乳特异性表达功能元件、参与抗旱诱导的MYB结合位点、TATA-box、SSⅡa基因起始密码子ATG。Figure 2. Sequence analysis of maize SSⅡa promoter表 1 SSⅡa启动子区顺式作用元件Table 1. Cis-acting elements of SSⅡa promoter元件名称 序列 元件功能 元件位置 CAAT-box CAATT、CAAAT、CAAT 启动子和增强子区常见的顺式作用元件 -2 380、-1 094、-2 088、-1 165、-1 377、-1 166、-1 095、-1 915、-2 013、-1 032、-815 CAT-box GCCACT 与分生组织表达相关的作用元件 -1 225、-546、-127 CE3 GACGCGTGTC 参与ABA和VP1响应的作用元件 -2 430 CG-motif CCATGGGG 光响应元件 -1 945 G-box CACATGG 光响应元件 -1 601、-392 I-box GGATAAGGTG 光响应元件 -2 419 MBS CGGTCA MYB结合位点 -2 452 Skn-1-motif GTCAT 胚乳表达所需的作用元件 -1 497、-1 087 GCN4-motif CAAGCCA 胚乳表达调控作用元件 -875 Sp1 CC(G/A)CCC 光响应元件 -1 279、-1 145 TATA-box TAATA、TATA、ATATAA、 核心启动子元件(在转录起始位点上游) -1 850、-2 028、-1 389、-1 847、-1 931、-1 848、-2 030、-529、-508 TC-rich repeats ATTTTCTCCA 参与防御和应激反应的作用元件 -1 815 TGA-element AACGAC 生长素响应的作用元件 -1 256、-247 TGACG-motif TGACG 参与茉莉酸甲酯响应的作用元件 -2 458、-1 198、-754 ABRE CACGTG 参与脱落酸响应的元件 -392 CGTCA-motif CGTCA 参与茉莉酸甲酯响应的作用元件 -213 GARE-motif TCTGTTG 赤霉素应答作用元件 -1 029 HSE AAAAAATTTC 参与热胁迫应答的作用元件 -824 MBS CAACTG 参与抗旱诱导的MYB结合位点 -1 000、-761 O2-site GATGATGTGG 参与玉米醇溶蛋白代谢调控的作用元件 -1 044 motif IIb CCGCCGCGCT 脱落酸响应元件 -796 2.2 SSⅡa启动子功能元件分析及构建缺失体设计
本研究成功克隆了玉米淀粉合成酶启动子SSⅡa的2 526 bp片段,并利用PlantCare软件对其进行分析[16],针对此片段上的顺式作用元件分别设计了5种5′缺失载体,其中:构建缺失体P1中含有8个常见顺式作用元件(CAAT),3个与分生组织表达相关的元件CAT-box,3个茉莉酸甲酯响应的元件CGTCA-motif和TGACG-motif,3个光响应元件GAG-motif和SP1,1个赤霉素应答元件GARE-motif,2个胚乳表达相关作用元件GCN4-motif和Skn-1 motif,1个热响应作用元件HSE,2个参与抗旱诱导的MYB结合位点MBS,1个参与玉米醇溶蛋白代谢调控的作用元件O2-site,2个TATA-box,2个生长素响应元件TGA-element,1个脱落酸响应元件motif IIb。
缺失体P2包括2个常见顺式作用元件(CAAT),2个与分生组织表达相关的元件CAT-box,2个茉莉酸甲酯响应的元件CGTCA-motif和TGACG-motif,1个光响应元件GAG-motif,1个热响应作用元件HSE,1个参与抗旱诱导的MYB结合位点MBS,2个TATA-box,1个生长素响应元件TGA-element,1个脱落酸响应元件motif IIb。
缺失体P3包括2个与分生组织表达相关的元件CAT-box,1个茉莉酸甲酯响应的元件CGTCA-motif,2个TATA-box,1个生长素响应元件TGA-element。
缺失体P4包括1个与分生组织表达相关的元件CAT-box,1个茉莉酸甲酯响应的元件CGTCA-motif,1个生长素响应元件TGA-element。
缺失体P5包括1个与分生组织表达相关的元件CAT-box,1个茉莉酸甲酯响应的元件CGTCA-motif,1个生长素响应元件TGA-element,P4与P5功能元件相同但P5与P4相比缺少预测到的转录起始位点(TSS)序列。
2.3 不同长度SSⅡa启动子5′缺失体植物表达载体构建
根据SSⅡa启动子分析结果,分别使用设计的缺失体引物进行PCR扩增,扩增结果如图 3A所示。采用EcoRⅠ、BglⅡ分别对5种不同长度SSⅡa启动子的PCR产物和植物表达载体pCAMBIA3301进行双酶切,分别构建了5个缺失体植物表达载体,转化大肠埃希菌感受态细胞后,以菌液为模板进行菌液PCR验证(图 3B),分别提取质粒进行双酶切验证,结果见图 3C。说明不同长度SSⅡa启动子植物表达载体构建成功,分别命名为:pCAMBIA-3301-P1、pCAMBIA-3301-P2、pCAMBIA-3301-P3、pCAMBIA-3301-P4和pCAMBIA-3301-P5,以下简称为P1、P2、P3、P4和P5。
![]() 图 3 不同长度SSⅡa启动子片段的扩增、重组载体PCR验证及双酶切验证结果M:DL2000 DNA Marker;1~5分别为缺失体P1、P2、P3、P4和P5。Figure 3. Amplification of SSⅡa promoter fragments of different length, PCR verification of recombinant vectors and double enzyme digestion verification
图 3 不同长度SSⅡa启动子片段的扩增、重组载体PCR验证及双酶切验证结果M:DL2000 DNA Marker;1~5分别为缺失体P1、P2、P3、P4和P5。Figure 3. Amplification of SSⅡa promoter fragments of different length, PCR verification of recombinant vectors and double enzyme digestion verification2.4 SSⅡa启动子在拟南芥中的功能分析
2.4.1 gus基因的PCR检测
用除草剂喷洒拟南芥,获得的抗性植株分别提取基因组DNA,以其为模板,gus基因为目标基因进行PCR检测,结果见图 4,由缺失体P1~P5均扩增出402 bp的条带,而非转基因对照的扩增结果为阴性,证明所采样的拟南芥均为成功转化不同长度启动子的阳性植株。
![]() 图 4 不同表达载体gus基因PCR扩增验证电泳结果M:DL2000 DNA Marker;1~2为缺失体P5,3~4为缺失体P4,5~6为缺失体P3,7~8为缺失体P2,9~10为缺失体P1;CK:非转基因对照。Figure 4. PCR detection results of gus gene in different expression vectors by electrophoresis
图 4 不同表达载体gus基因PCR扩增验证电泳结果M:DL2000 DNA Marker;1~2为缺失体P5,3~4为缺失体P4,5~6为缺失体P3,7~8为缺失体P2,9~10为缺失体P1;CK:非转基因对照。Figure 4. PCR detection results of gus gene in different expression vectors by electrophoresis2.4.2 转基因植株的GUS组织化学分析
在成熟期阶段,分别采取叶片和果荚为试验材料进行GUS染色,结果见图 5,野生型拟南芥(CK)叶片未见蓝色,5种类型不同转启动子的拟南芥叶片均呈现不均匀蓝色;野生型拟南芥果荚未见蓝色,转基因拟南芥果荚均有蓝色,且不同长度启动子的果荚,蓝色深浅不同,P1和P2果荚染色较深,P3、P4和P5果荚染色较浅。
![]() 图 5 成熟期转基因拟南芥植株叶片和果荚GUS组织化学染色Figure 5. Histochemical analysis of GUS activities in leaves and pods of transgenic Arabidopsis thaliana plant at maturity
图 5 成熟期转基因拟南芥植株叶片和果荚GUS组织化学染色Figure 5. Histochemical analysis of GUS activities in leaves and pods of transgenic Arabidopsis thaliana plant at maturity2.4.3 gus基因的定量分析
转基因植株经组织化学染色分析后,可以初步判断启动子驱动的gus基因的表达部位和大致表达强度,但并不能判断启动子的活力大小,为了进一步明确5种启动子的活性,利用荧光定量PCR技术从转录水平上检测各启动子驱动的gus基因的表达情况,图 6所示为成熟期5种转基因植株叶片和种子gus基因相对表达量。由图 6可知,野生型拟南芥(CK)叶片和种子中gus基因均不表达,这与GUS染色结果一致。5种转基因叶片中,gus基因表达量P1最高,其他基本一致。种子中gus基因P1和P2表达量相近,高于P3、P4和P5,P5表达量最低。P1和P2中含有胚乳特异性表达的功能元件。本研究结果表明,缺失胚乳表达功能元件可导致所驱动的gus基因在种子中表达量下降(P3、P4和P5)。SSⅡa启动子具有的胚乳特异性,种子gus基因表达量差异可能与功能元件的数量有关[16]。
![]() 图 6 成熟期转基因拟南芥叶片和种子中gus基因的相对表达量Figure 6. Relative expression levels of gus gene in leaves and seeds of transgenic Arabidopsis thaliana plant at maturity
图 6 成熟期转基因拟南芥叶片和种子中gus基因的相对表达量Figure 6. Relative expression levels of gus gene in leaves and seeds of transgenic Arabidopsis thaliana plant at maturity3. 讨论与结论
2009年玉米基因组测序工作的完成,为玉米功能基因及启动子序列的克隆带来了极大的方便。本试验利用玉米淀粉合成酶基因SSⅡa核酸序列,BLAST分析出SSⅡa 5′上游DNA序列,设计特异引物,利用常规PCR方法克隆了2 526 bp SSⅡa基因5′侧翼序列,与NCBI中公布的玉米基因组序列比对后,一致性为99.96%。
目前,对玉米SSⅡa启动子的研究报道较少[17],但Harn等[8]1998年最先从玉米胚乳中成功的克隆了zmSSⅡa和zmSSⅡb的cDNA,SSⅡa在玉米淀粉代谢网络中也主要在胚乳中行使功能,参与支链淀粉的合成;任红丽等[18]与Hu等[19]分别鉴定了玉米可溶性淀粉合成酶基因的SSⅠ和SSⅢ启动子均为胚乳特异性启动子;Li等[20]研究表明籼稻SSⅡa启动子为胚乳特异性启动子。以上研究结果预示本研究克隆的玉米SSⅡa启动子可能具有胚乳特异性。
本研究结果经PlantCare在线分析软件分析,发现其除具有典型启动子基本顺式作用元件之外,还含有胚乳特异性表达功能元件,抗旱诱导结合位点,光诱导元件等胁迫诱导顺式作用元件。为了研究部分顺式作用元件的功能,构建了5个缺失体载体,主要研究胚乳特异性功能元件、抗旱诱导MYB结合位点等的功能。重新设计5种类型的引物,扩增SSⅡa启动子序列的长度分别为1 407 bp(P1)、867 bp(P2)、633 bp(P3)、483 bp(P4)和365 bp(P5),用于构建5个不同长度SSⅡa启动子缺失的植物表达载体,分别转化拟南芥,通过基因组PCR检测,均扩增出402 bp的条带,可证明所采样的拟南芥均为成功转化不同长度启动子的阳性植株。利用GUS化学法对转基因阳性拟南芥成熟期叶片和果荚进行染色,初步验证了gus基因表达部位和大致表达强度;通过荧光定量技术对转基因阳性拟南芥成熟期叶片和种子进行gus基因定量表达,证明了长度为1 407 bp(P1)和867 bp(P2)的SSⅡa启动子具有胚乳特异性。
本研究对5种启动子的功能比较分析只是初步结果,利用P1和P2联合考察胚乳特异性功能元件的作用,对其功能的深入探讨,如利用P2和P3联合考察抗旱诱导MYB结合位点的作用,利用P3和P4联合考察启动子中TATA序列是否为此启动子行使功能的TATA-box等功能的深入研究,还有待后续实验验证。通过对以上5种启动子缺失体功能的进一步研究,期望能够明确SSⅡa启动子中部分顺式作用元件的功能,为SSⅡa启动子的应用提供理论依据,为组织特异性启动子的开发和应用提供候选启动子,为玉米淀粉代谢网络调控的研究提供基础。
-
![]()
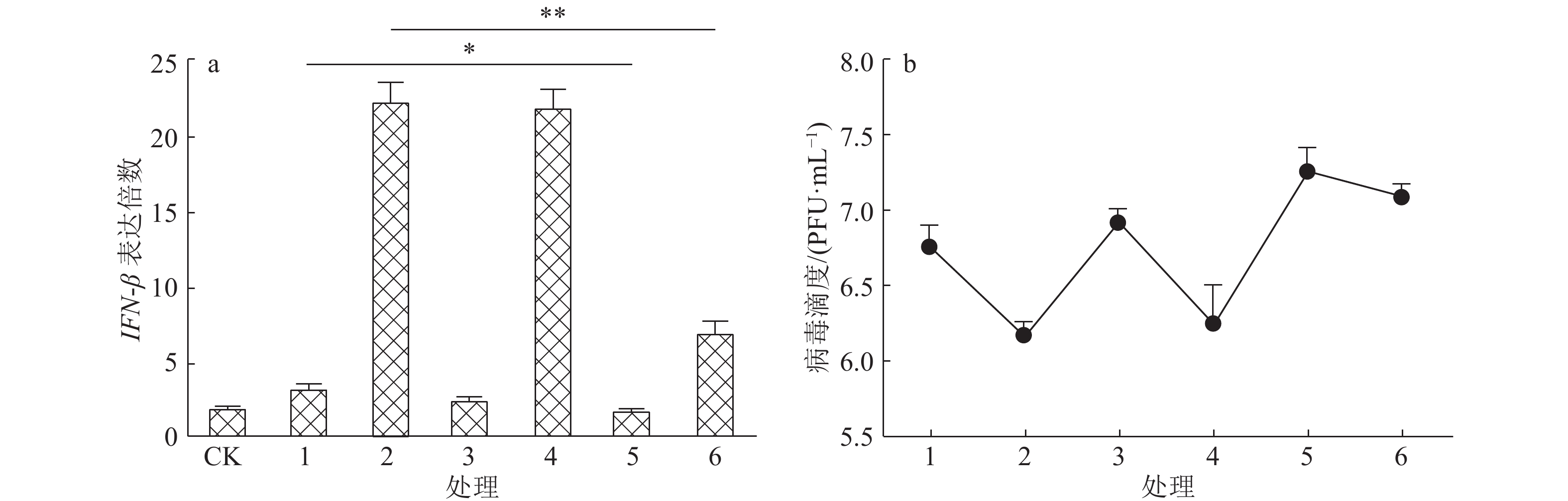
图 1 Hep-dG感染NA细胞激活IFN-β信号通路
a: Hep-dG感染增强IFN-β基因的表达水平,图中“*”、“**”和“***”分别表示直线两端所对应的样本差异达0.05、0.01和0.001的显著水平(t检验);b:Hep-dG感染刺激STAT1的表达和激活,1:空白对照,2:rHep-Flury,3:Hep-dG
Figure 1. Hep-dG infection activated the IFN-β signaling pathway in NA cells
![]()
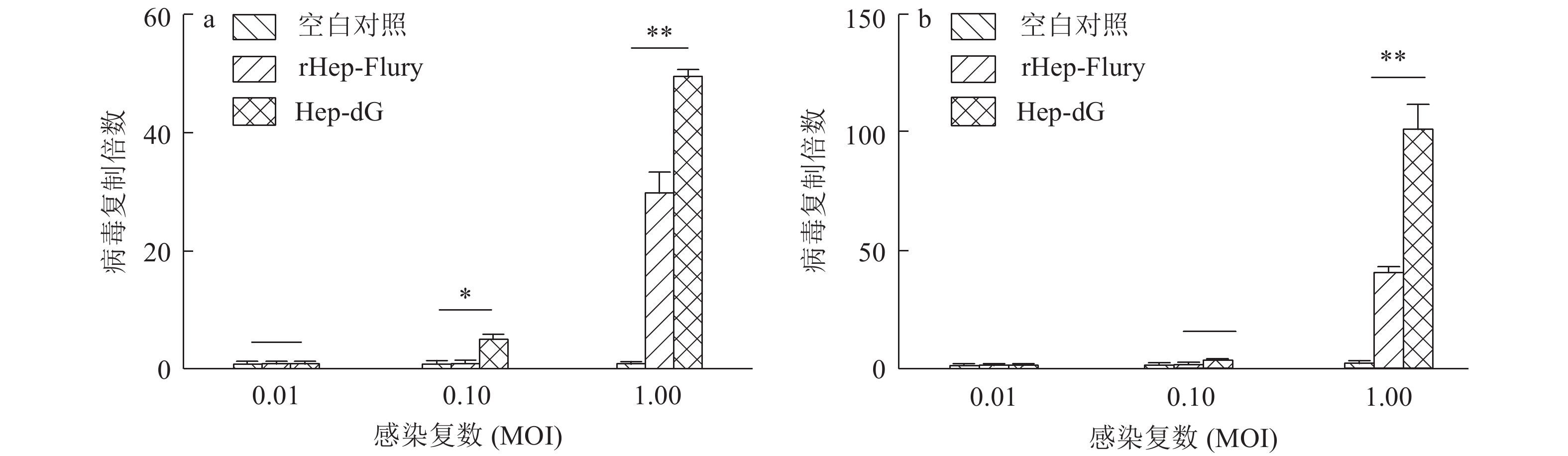
图 2 狂犬病病毒吸附和入侵NA细胞的分析
a:细胞表面吸附狂犬病病毒的分析;b:狂犬病病毒入侵NA细胞的分析,各图中“*”和“**”分别表示直线两端所对应的样本差异达0.05和0.01的显著水平(t检验)
Figure 2. Analyses of RABVs adsorbing and invading NA cells under different infection multiplicities
![]()
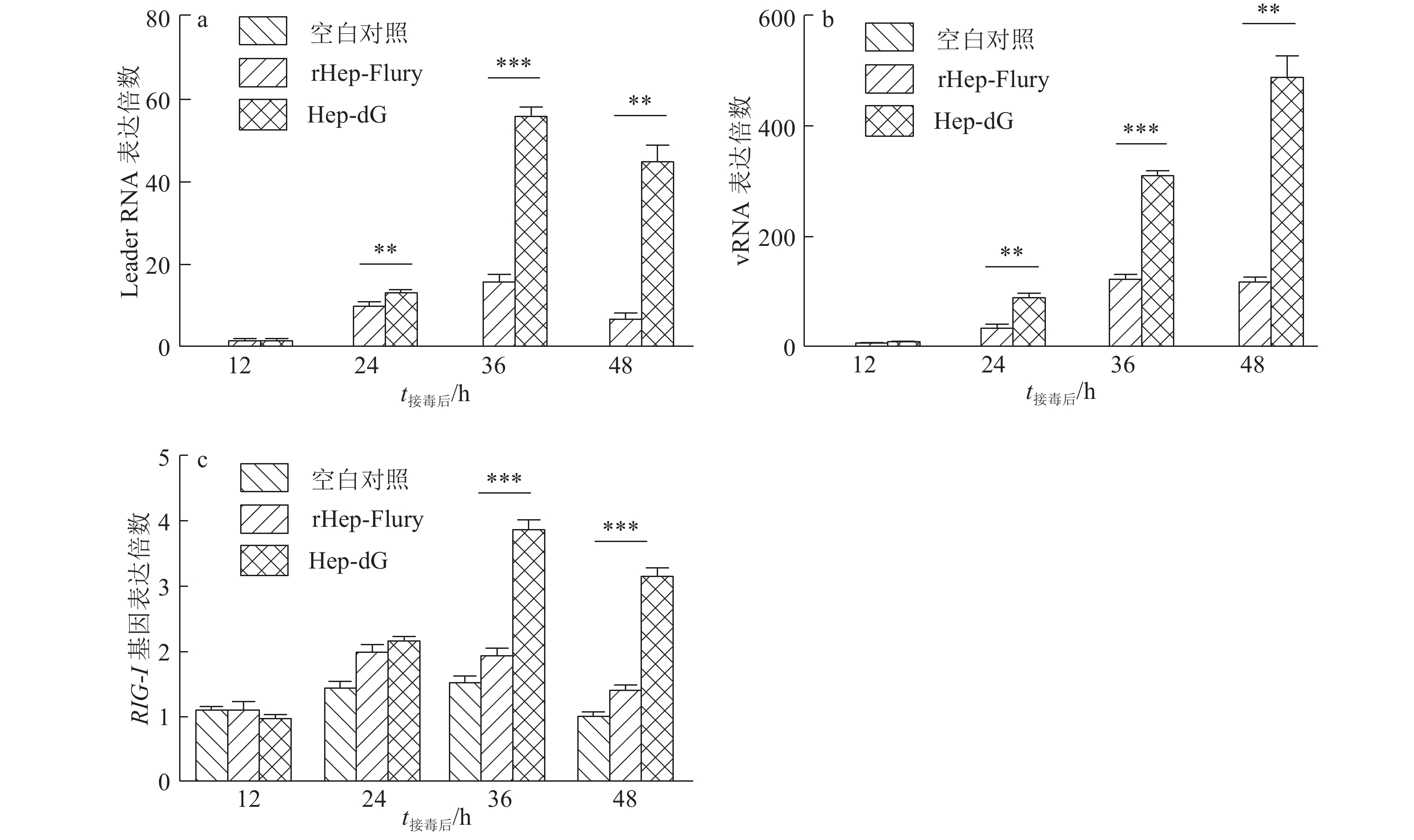
图 3 狂犬病病毒感染对Leader RNA、vRNA和RIG-I基因表达的影响
a:Leader RNA的表达,b:vRNA的表达,c:RIG-I基因的表达;各图中“**”和“***”分别表示直线两端所对应的样本差异达0.01和0.001的显著水平(t检验)
Figure 3. Effects of rabies virus infections on expressions of Leader RNA,vRNA and RIG-Igene
![]()
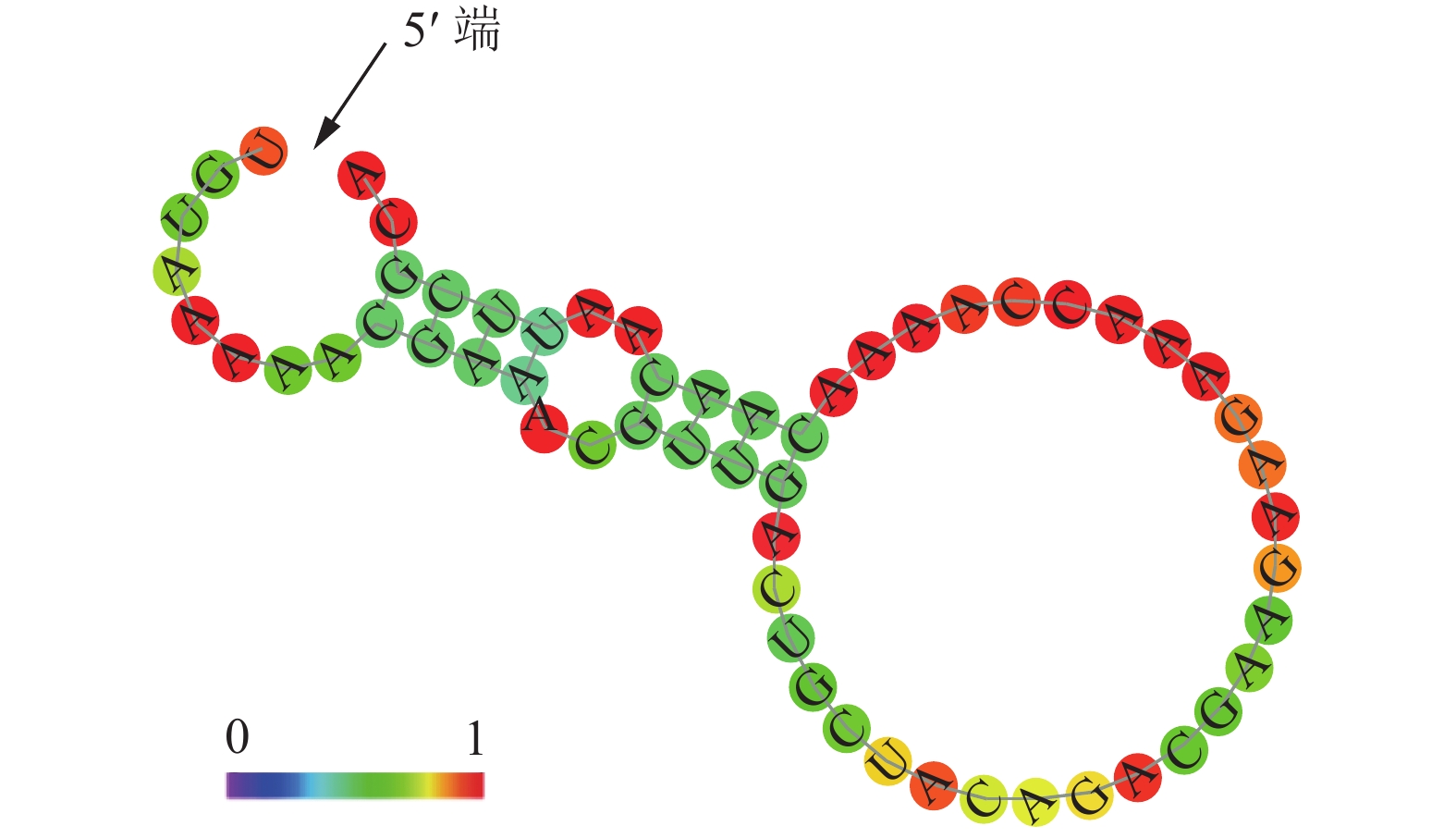
图 4 基于最小自由能的狂犬病病毒rHep-Flury和Hep-dG Leader RNA的二级结构
Figure 4. The secondary structure of rabies viruses rHep-Flury and Hep-dG Leader RNA based on the minimum free energy
![]()
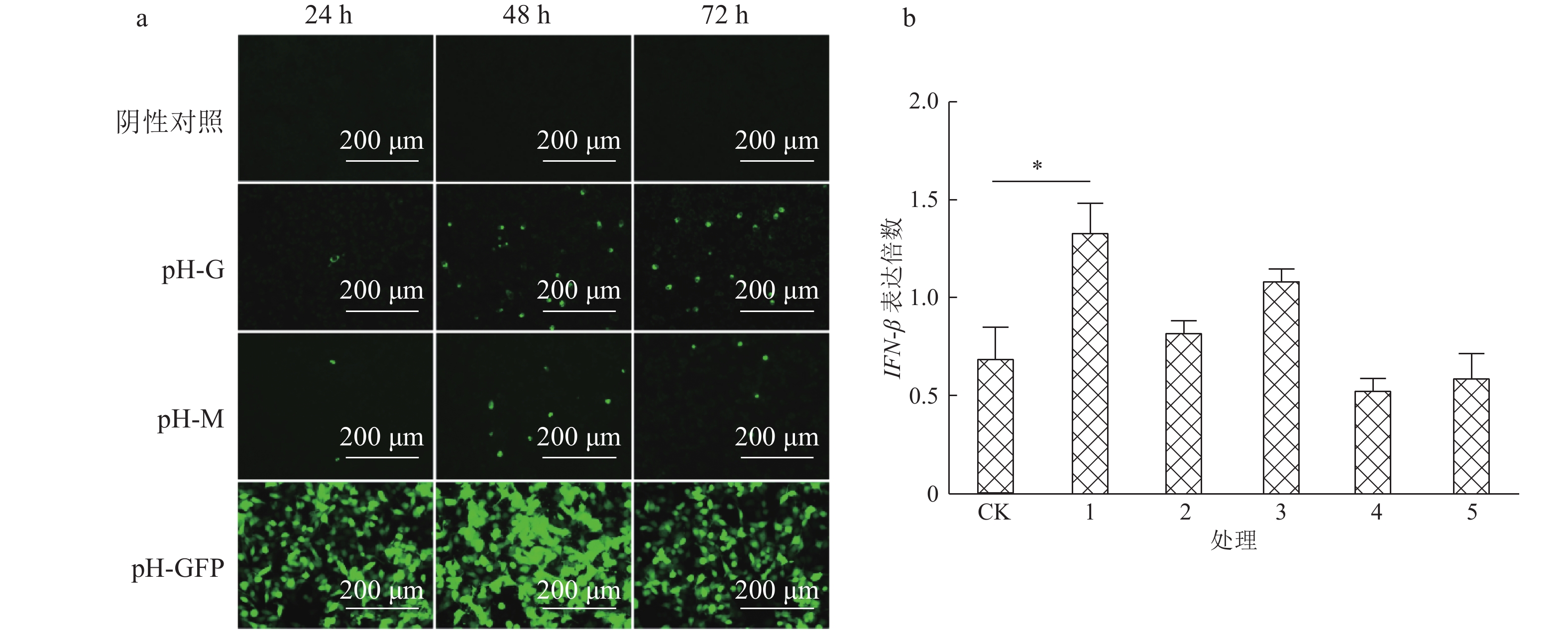
图 5 G蛋白的真核表达对NA细胞中IFN-β转录的影响
a:pH-G、pH-M和pH-GFP的荧光表达;b:IFN-β基因的表达,CK:阴性对照,1:5.0 μg的pH-G,2:2.5 μg的pH-G,3:5.0 μg的pH-M,4:2.5 μg的pH-M,5:阳性对照,图中“*”表示直线两端所对应的样本达0.05的显著水平(t检验)
Figure 5. Effect of G protein eukaryotic expression on IFN-β transcription in NA cell
![]()
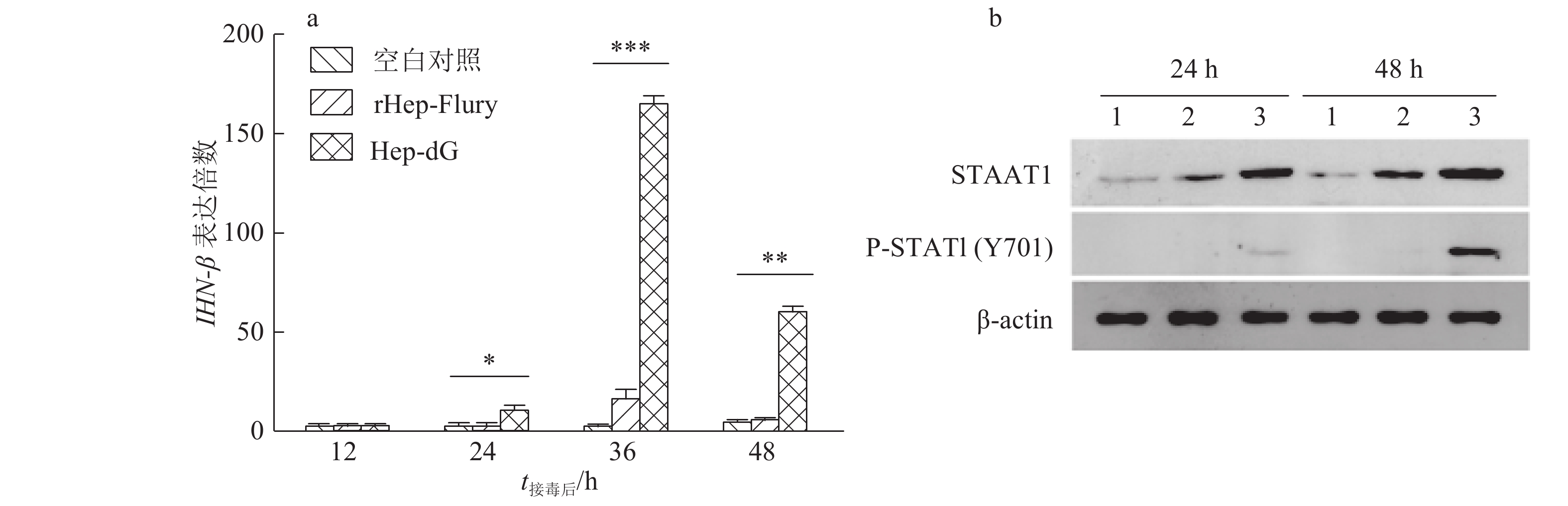
图 6 阻断IFN-β通路对狂犬病病毒复制的影响
a.抗体阻断IFN-β的表达,CK:空白对照,1:rHep-Flury,2:Hep-dG,3:rHep-Flury+IgG,4:Hep-dG+IgG,5:rHep-Flury+Anti,6:Hep-dG+Anti,图中“*”和“**”分别表示直线两端所对应的样本达0.05和0.01的显著水平(t检验);b.病毒滴度的检测,1:rHep-Flury,2:Hep-dG,3:rHep-Flury+IgG,4:Hep-dG+IgG,5:rHep-Flury+Anti,6:Hep-dG+Anti
Figure 6. Effect of blocking IFN-β pathway on rabies virus replication
表 1 荧光定量PCR检测引物
Table 1 Primers used for fluorescence quantitative PCR detection
被检基因 引物序列 引物名称 产物长度/bp Leader RNA Leader-F 5'-CCAGATGCTTGGCGTCCT-3' 67 Leader-R 5'-ACGCTTAACAACAAAACC-3' vRNA vRNA-F 5'-GGAAAAGGGACATTTGAAAGAA-3' 130 vRNA-R 5'-AGTCCTCGTCATCGGAGTTGAC-3' RIG-I RIGI-F 5'-GCAAGTGCTTCCTCCTGACC-3' 142 RIGI-R 5'-ATGCGGTGAACCGTCTTTCC-3' GAPDH GAPDH-F 5'-AGAGTGTTTCCTCGTCCCGT-3' 199 GAPDH-R 5'-CTGTGCCGTTGAATTTGCCG-3'  下载: 导出CSV
下载: 导出CSV
-
[1] 王朝, 周明, 傅振芳, 等. 狂犬病病毒逃逸宿主天然免疫反应的研究进展[J]. 生命科学, 2017(3): 237-244. [2] 张旭, 纪森林, 赵雯, 等. 狂犬病病毒致病机制的最新研究进展[J]. 中国兽医科学, 2018, 48(3): 295-304. [3] MEBATSION T, KONIG M, CONZELMANN K K. Budding of rabies virus particles in the absence of the spike glycoprotein[J]. Cell, 1996, 84(6): 941-951.
[4] MEBATSION T, WEILAND F, CONZELMANN K K. Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G[J]. J Virol, 1999, 73(1): 242-250.
[5] MACFARLAN R I, DIETZSCHOLD B, WIKTOR T J, et al. T cell responses to cleaved rabies virus glycoprotein and to synthetic peptides[J]. J Immunol, 1984, 133(5): 2748-2752.
[6] 汪孟航, 朱洪伟, 何民辉, 等. 狂犬病病毒糖蛋白生物学功能研究进展[J]. 中国畜牧兽医, 2016(12): 3349-3355. [7] FISHER C R, STREICKER D G, SCHNELL M J. The spread and evolution of rabies virus: Conquering new frontiers[J]. Nat Rev Microbiol, 2018, 16(4): 241-255.
[8] LIU X, YANG Y, SUN Z, et al. A recombinant rabies virus encoding two copies of the glycoprotein gene confers protection in dogs against a virulent challenge[J]. PLoS One, 2014, 9(2): e87105.
[9] PREHAUD C, LAY S, DIETZSCHOLD B, et al. Glycoprotein of nonpathogenic rabies viruses is a key determinant of human cell apoptosis[J]. J Virol, 2003, 77(19): 10537-10547.
[10] FABER M, PULMANAUSAHAKUL R, HODAWADEKAR S S, et al. Overexpression of the rabies virus glycoprotein results in enhancement of apoptosis and antiviral immune response[J]. J Virol, 2002, 76(7): 3374-3381.
[11] YANG Y, LIU W, YAN G, et al. iTRAQ protein profile analysis of neuroblastoma (NA) cells infected with the rabies viruses rHep-Flury and Hep-dG[J/OL]. Front Microbiol, 2015, 6: 691. doi: 10.3389/fmicb.2015.00691.
[12] HABJAN M, PENSKI N, SPIEGEL M, et al. T7 RNA polymerase-dependent and -independent systems for cDNA-based rescue of Rift Valley fever virus[J]. J Gen Virol, 2008, 89(9): 2157-2166.
[13] YANG Y, HUANG Y, GNANADURAI C W, et al. The inability of wild-type rabies virus to activate dendritic cells is dependent on the glycoprotein and correlates with its low level of the de novo-synthesized leader RNA[J]. J Virol, 2015, 89(4): 2157-2169.
[14] WU J, MA C, WANG H, et al. A MyD88-JAK1-STAT1 complex directly induces SOCS-1 expression in macrophages infected with Group A Streptococcus[J]. Cell Mol Immunol, 2015, 12(3): 373-383.
[15] OGINO T, YADAV S P, BANERJEE A K. Histidine-mediated RNA transfer to GDP for unique mRNA capping by vesicular stomatitis virus RNA polymerase[J]. Proc Natl Acad Sci USA, 2010, 107(8): 3463-3468.
[16] GERLIER D, LYLES D S. Interplay between innate immunity and negative-strand RNA viruses: Towards a rational model[J]. Microbiol Mol Biol Rev, 2011, 75(3): 468-490.
[17] MOYER S A, ABRAHAM G, ADLER R, et al. Methylated and blocked 5' termini in vesicular stomatitis virus in vivo mRNAs[J]. Cell, 1975, 5(1): 59-67.
[18] BRZOZKA K, FINKE S, CONZELMANN K K. Identification of the rabies virus alpha/beta interferon antagonist: Phosphoprotein P interferes with phosphorylation of interferon regulatory factor 3[J]. J Virol, 2005, 79(12): 7673-7681.
[19] RIEDER M, CONZELMANN K K. Interferon in rabies virus infection[J]. Adv Virus Res, 2011, 79: 91-114.
[20] 王林栋, 张守峰, 刘晔, 等. 基于RIG-I的狂犬病病毒免疫逃逸机制[J]. 中国生物制品学杂志, 2013(10): 1517-1521. [21] VIDY A, CHELBI-ALIX M, BLONDEL D. Rabies virus P protein interacts with STAT1 and inhibits interferon signal transduction pathways[J]. J Virol, 2005, 79(22): 14411-14420.
[22] VIDY A, EL B J, CHELBI-ALIX M K, et al. The nucleocytoplasmic rabies virus P protein counteracts interferon signaling by inhibiting both nuclear accumulation and DNA binding of STAT1[J]. J Virol, 2007, 81(8): 4255-4263.
[23] PLUMET S, HERSCHKE F, BOURHIS J M, et al. Cytosolic 5'-triphosphate ended viral leader transcript of measles virus as activator of the RIG I-mediated interferon response[J]. PLoS One, 2007, 2(3): e279.
[24] FAUL E J, WANJALLA C N, SUTHAR M S, et al. Rabies virus infection induces type I interferon production in an IPS-1 dependent manner while dendritic cell activation relies on IFNAR signaling[J]. PLoS Pathog, 2010, 6(7): e1001016.
[25] KATZ I, GUEDES F, FERNANDES E R, et al. Immunological aspects of rabies: A literature review[J]. Arch Virol, 2017, 162(11): 3251-3268.






计量
- 文章访问数: 1670
- HTML全文浏览量: 2
- PDF下载量: 1866
