
Prokaryotic expression and activity detection of bacteriophage lysin Cp51 against Clostridium perfringens type A
-
摘要:目的
原核表达A型产气荚膜梭菌噬菌体裂解酶Cp51并研究其对A型产气荚膜梭菌Clostridium perfringens的体外抗菌活性。
方法合成噬菌体裂解酶Cp51基因,并构建了pET-32a-Cp51原核表达载体,转化到大肠埃希菌Escherichia coli BL21(DE3),经终浓度为0.5 mmol·L–1的IPTG诱导以及镍柱纯化后,获得了可溶性的Cp51重组蛋白;用比浊法检测Cp51重组蛋白的杀菌活性。
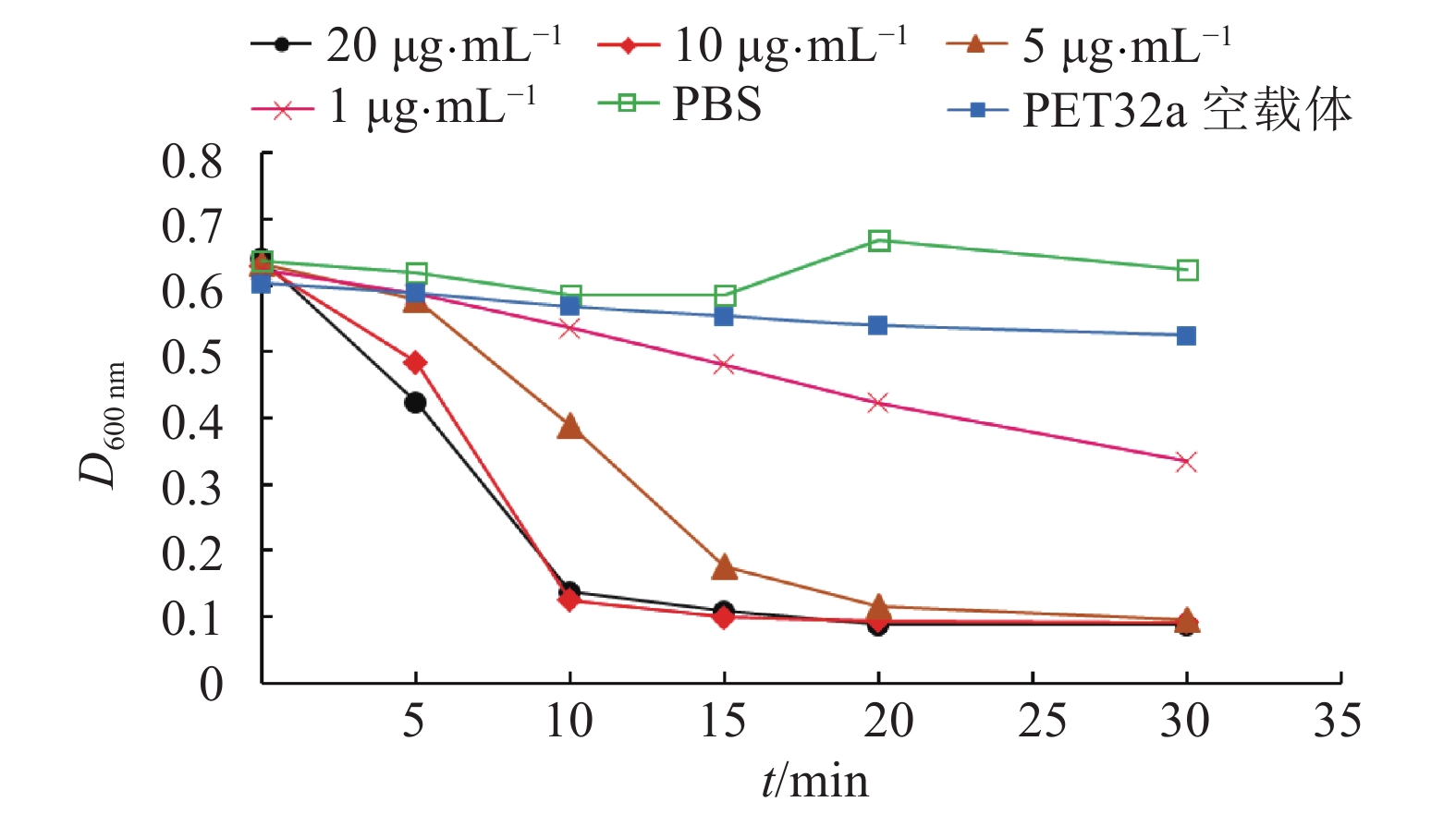
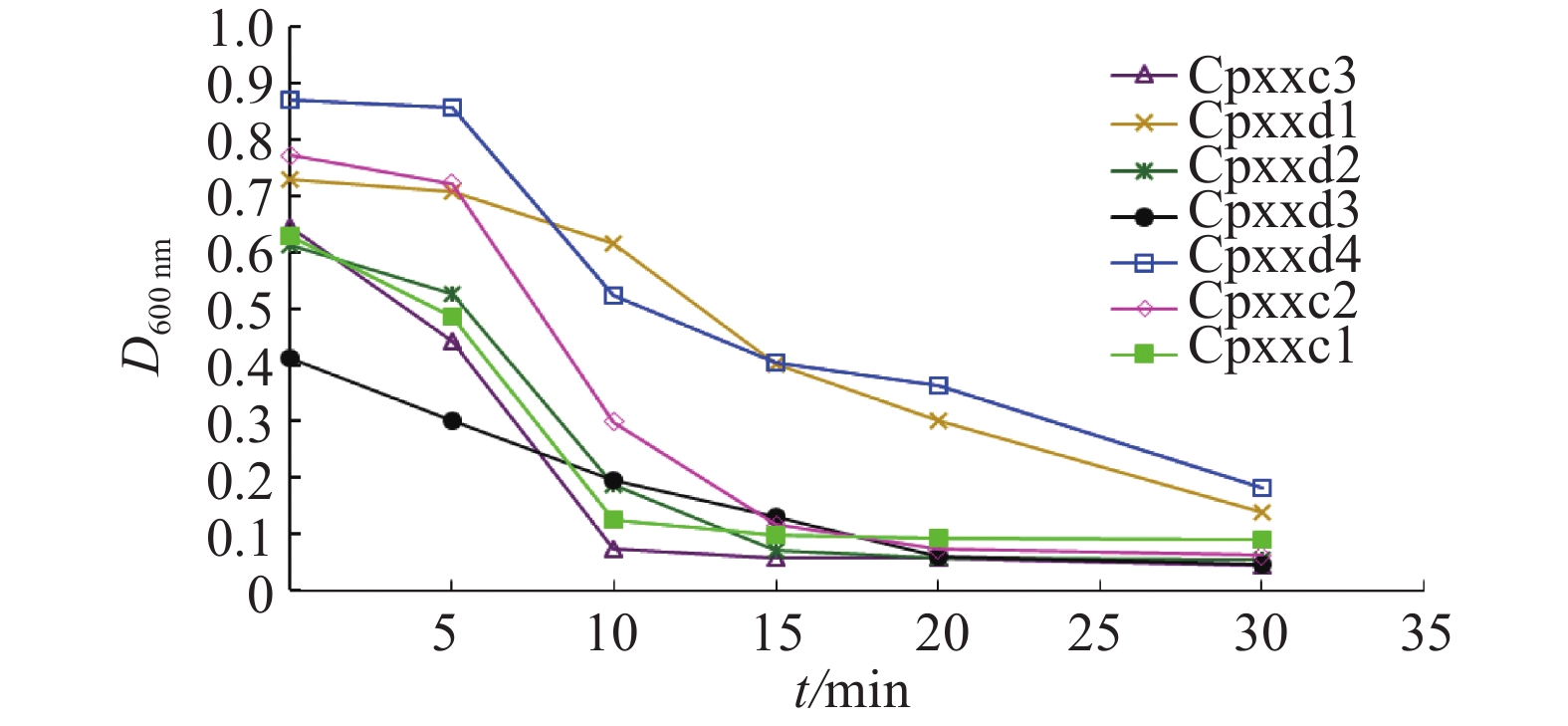
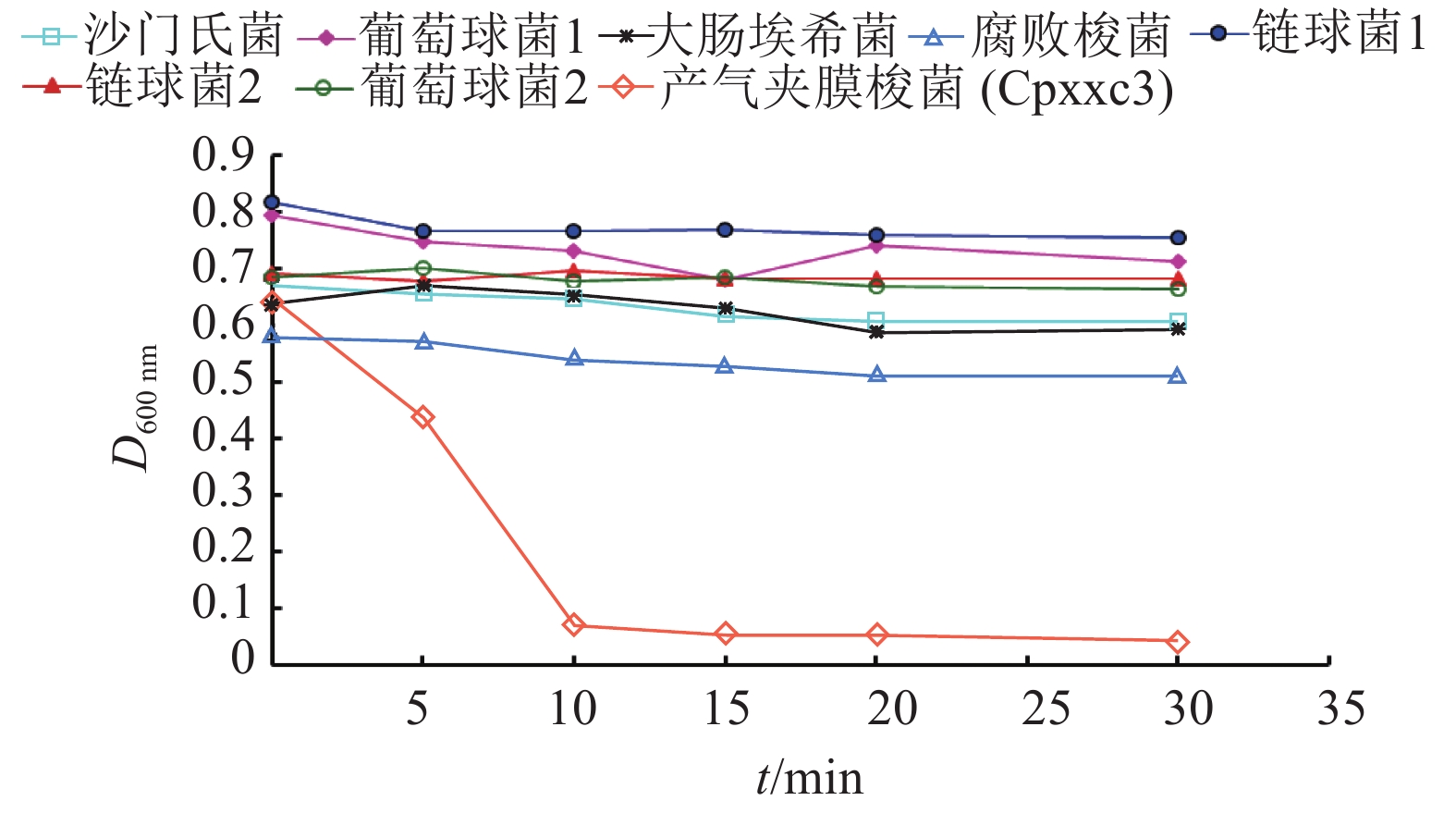
结果噬菌体裂解酶Cp51重组蛋白能够有效降低7株A型产气荚膜梭菌的浊度,裂解酶质量浓度在5 μg·mL–1以上、作用30 min对A型产气荚膜梭菌的杀菌率可达到99.99%以上,而对其他种类细菌无杀菌效果。
结论A型产气荚膜梭菌噬菌体裂解酶Cp51重组蛋白对A型产气荚膜梭菌有较强的体外杀菌活性和特异性,研究结果为后续裂解酶Cp51的临床应用奠定基础。
Abstract:ObjectiveTo construct a prokaryotic expression system of type A Clostridium perfringens phage lysin Cp51, and study its antibacterial activity against C. Perfringens type A in vitro.
MethodBacteriophage lysin Cp51 gene was synthesized. The prokaryotic expression vector pET-32a-Cp51 was constructed and transformed into Escherichia coli BL21(DE3). After induction using 0.5 mmol·L–1 IPTG, the soluble recombinant protein Cp51 was successfully expressed, and was subsequently purified with Ni2+-NTA affinity chromatography. The antibacterial activity of the recombinant protein Cp51 was detected by kinetic turbidimetric assay.
ResultThe bacteria turbidities of seven strains of C. perfringens type A were effectively reduced by the recombinant protein Cp51. The bactericidal rate was above 99.99% in 30 min after treatment of recombinant protein Cp51 at the concentration of above 5 μg·mL–1. The Cp51 protein had no bactericidal effect against other types of bacteria.
ConclusionThe recombinant protein of bacteriophage lysine Cp51 has strong bactericidal activity and specificity in vitro against type A C. perfringens, which could provide a basis for clinical application of Cp51 lysin.
-
Keywords:
- Clostridium perfringens /
- bacteriophage /
- lysin /
- protein expression /
- bactericidal activity
-
高效、精准的基因组编辑修饰是理解和探索重要生物学问题的基础方法,同时也是精准育种、基因治疗等的重要手段,对农业及医疗领域的发展具有战略性意义。随着生物技术的不断发展,科学家先后研发出锌指核酸酶(Zinc-finger nucleases,ZFN);类转录激活因子样效应物核酸酶(Transcription activator-like effector nucleases,TALEN);CRISPR/Cas系统(Clustered regularly interspaced short palindromic repeats-associated proteins system)等一系列重要的序列特异性核酸酶(Sequence specific nucleases,SSNs);实现了基因组目标位点的精准识别和切割[1-2]。其中,CRISPR/Cas系统[3-5]以高效、简单、低成本等优点迅速成为应用场景最广的基因组编辑工具,并广泛应用于基础研究、医疗和育种[6-9]。
通常情况下,以SSNs为基础的基因编辑技术可以实现高效的定点切割,激活细胞体内的非同源末端连接(Non-homologous end joining,NHEJ)或同源重组(Homologous recombination,HR)修复途径,实现目标位点序列的改变[10]。然而,NHEJ的修复结果通常会产生少量碱基的插入或缺失(Insertions/deletions,Indels),难以产生预期的编辑类型。而通过HR修复虽然可以实现精确碱基或片段的靶向置换,但其低效率仍然是许多高等生物例如植物中难以克服的瓶颈问题[11]。为此,美国哈佛大学Liu David课题组于2016年开发出碱基编辑的全新技术体系,运用胞嘧啶碱基编辑器(Cytosine base editor,CBE)[12]及腺嘌呤碱基编辑器(Adenosine base editor,ABE)[13],采用缺口酶形式的Cas9(Nickase Cas9,nCas9)绕开NHEJ及HR修复,通过单链断裂修复途径,成功实现了嘌呤间和嘧啶间的碱基转换(Base transition),同时极大减少了双链修复过程中可能会存在的非预期的Indels[14-17]。然而,科学家们也发现,碱基编辑技术存在大量全基因组及转录水平上难以预测的随机脱靶事件。这主要是由于脱氨酶非特异性识别DNA或RNA链上的碱基并诱发该碱基发生脱氨所导致,在应用层面造成严重的安全隐患[18-21]。此外,常规的碱基编辑技术只能实现碱基之间的转换,不能实现碱基间的颠换(Base transversion),也不能实现碱基的精准增删[11]。虽然科学家们开发了一系列碱基颠换新工具,但他们的效率和产物纯度与上述的碱基编辑器相比仍存在较大的差距[22-25]。
由于碱基编辑系统存在上述缺陷,2019年,Liu David课题组成功研发出引导编辑器(Prime editor)[26],借助全新的技术路径完成了任意形式的碱基替换,从而再一次将基因编辑技术的精准度推向新的高度。该系统克服了碱基编辑只能实现4种碱基转换的问题,可以完成其余8种形式的碱基颠换以及小片段碱基的精准插入和删除[26-27]。自引导编辑(Prime editing,PE)开发以来就广受关注,并被应用在医疗、农业等多个研究领域[28-29]。然而,初代的PE仍存在如效率偏低、安全性尚未全面评估、难以实现大片段操纵等诸多问题需要解决。目前,学者们围绕PE存在的问题进行了大量的优化和研究,取得了一系列重要的进展。本文将概括PE的组成和技术原理,阐述PE存在的局限性,并综述和总结当前研究学者所采取的相关优化措施,最后对该系统目前及今后的应用方向和发展前景进行展望。
1. PE系统的工作原理
通常情况下,引导编辑器主要由2个关键元件组成:H840A缺口酶形式的nCas9融合逆转录酶(Reverse transcriptase,RT)组成的融合蛋白和引导编辑向导RNA (Prime editing guide RNA,pegRNA)(图1)。pegRNA与常规的sgRNA非常类似,但其3'端延伸出一段包含PBS (Primer binding site)序列和RT模板(RT template,RTT)的序列。这2个关键元件会形成复合物,结合至目标位点。其中,融合蛋白中的nCas9主要行使非靶标链的切割功能,暴露出非靶标链上的单链。此时,pegRNA上的PBS序列可以与该暴露出来的DNA单链结合,启动PE过程。在RT的作用下,pegRNA上的RTT序列信息被逆转录成DNA形式,留在DNA断裂处,形成被称为3' flap的DNA单链结构。对于3' flap上的DNA信息如何被引入基因组目前尚不清楚,可能性最大的猜测是3' flap与5' flap之间存在动态转换,使所需DNA信息有机会与基因组的靶标链结合,之后5' flap会在细胞修复的过程中被切除,经过DNA修复过程,最终实现基因组信息的修改(图1)。在这个过程中,融合蛋白承担了切割目标位点非靶标链和逆转录的双重功能,而pegRNA既引导PE识别目标位点,又包含了编辑所需的信息。通过这2个组分,PE系统实现了识别、切割、起始逆转录的引物序列结合、逆转录等一系列过程,并将所需DNA信息直接逆转录至目标位点的断裂处[26]。PE系统的设计非常简单精巧,无需引入DNA模板,也不产生双链断裂,是一种非常聪明的技术策略。
![]() 图 1 引导编辑器的构成及引导编辑的原理示意图左侧为引导编辑器的构成,右侧为flap的转换及DNA修复过程;其中,PE3和PE3b系统需要额外的nicking sgRNA产生缺口,而PE2则不需要Figure 1. Schematic diagram of prime editor and desired prime edit installingLeft panel: Diagram of prime editor, right panel: Flap transition and DNA repair process; The PE3 and PE3b systems use an additional nicking sgRNA to generate a nick on DNA compared to PE2
图 1 引导编辑器的构成及引导编辑的原理示意图左侧为引导编辑器的构成,右侧为flap的转换及DNA修复过程;其中,PE3和PE3b系统需要额外的nicking sgRNA产生缺口,而PE2则不需要Figure 1. Schematic diagram of prime editor and desired prime edit installingLeft panel: Diagram of prime editor, right panel: Flap transition and DNA repair process; The PE3 and PE3b systems use an additional nicking sgRNA to generate a nick on DNA compared to PE2为了进一步提升PE系统的性能,Liu David团队接连开发了3个优化的PE版本,即:PE2、PE3、PE3b,其中,PE3和PE3b通过在系统中引入nicking sgRNA,使PE识别并切割编辑链的对侧链,从而促进修复机制保留编辑链上的编辑信息(图1)。PE3b通过将nicking sgRNA设计为只能识别突变引入后的序列,从而将编辑链切割和对侧链切割2个过程从时间上分开,减小双链同时断裂的可能性,从而大大降低非预期的Indels事件[26]。
PE系统被开发之后就有多个团队将其与常规HR技术及碱基编辑技术进行对比。例如Liu David 团队在开发该系统时就发现PE系统编辑效率相当或更高于HR技术和碱基编辑技术,并且相比之下副产物比例更低,在编辑产物纯度上更具优势[26]。Gao等[30]也在小鼠中获得了类似的结论;Lin等[31]在植物细胞中将PE系统与碱基编辑系统进行比较,也发现PE在精确编辑上更具优势。因此,PE系统是一个极具开发潜力、具有独特优势的全新技术。
2. PE系统性能的限制因素
尽管PE系统与其他技术相比具有优势和前景,但依然存在诸多问题需要进一步解决完善。尤其是在该技术的开发初期,其编辑效率仍不理想。此外,可能受RT逆转录效率或DNA修复效率等因素的影响,PE系统难以实现DNA大片段的基因组操纵。在PE系统的安全性方面,也亟待全面、详细的评估。因此,利用PE系统还难以做到真正意义上的高效、精确、稳定、安全的编辑,它还具备较大的优化空间和潜力。我们从以下几个方面归纳了目前其存在的主要问题,并对其产生原因及影响进行阐述。
2.1 效率偏低
多个团队的测试结果表明,PE系统在一些位点上工作效率不够理想,甚至在有些物种中获得精准编辑的频率非常低,难以真正满足研究及应用层面的需求[31-33]。由于PE系统的修复过程较为复杂,涉及PE对目标位点的识别和结合、PBS序列与单链序列的结合、RTT的逆转录、flap结构引入基因组等一系列过程,每一个步骤都会对PE系统的编辑效率产生影响。例如,Vu等[34]发现PE系统的pegRNA自身会形成二级结构,影响其识别基因组目标位点的能力。Lin等[31]发现不同活性的RT会对PE效率有直接影响,证明RT模板的逆转录过程对PE十分重要。因此,需要针对以上过程中所参与的元件或反应进行优化,提升PE系统的工作效率。
2.2 难以实现大片段操纵
根据报道,由基因的插入、重复和缺失所引起的人类致病性变体已占到已知人类致病性变体的14%左右,而且,许多基因组异常都涉及较大的DNA片段(>100 bp)变异[35]。虽然Anzalone等[26]给出了运用PE系统产生包括高达44 bp的小片段精准插入和高达80 bp的小片段精准删除的例子,但仅限于少量细胞类型的测试,并且没有进行更大片段编辑的尝试。Lin等[31]在植物细胞中测试发现PE的编辑效率随着所需插入或删除长度的增长而显著降低,能检测到最长30 nt的插入和40 nt的删除,但其效率分别只有0.3%和0.7%。Wang等[36]通过PE实现标签插入的效果也并不理想,编辑效率低于0.1%。通过常规PE实现大片段插入、删除或替换的成功例子也很少。因此,对于初代的PE系统而言,大片段操纵仍具有较大的挑战性。
2.3 编辑范围受限
PE系统的编辑范围受编辑窗口大小影响。虽然理论上编辑窗口可以远离切割位置,但受RT活性、RTT长度、细胞修复机制等因素的影响,过远的编辑位置通常效率很低,这一点在多个团队的研究结果中均得到验证[31-32, 37-39],这一现象也从侧面反映了PE系统效率仍然不足。此外,初代的PE系统通常使用化脓性链球菌Streptococcus pyogenes Cas9 (SpCas9)进行编辑,其编辑需要较为严格的间隔序列前体临近基序(Protospacer adjacent motif,PAM),也对编辑范围产生了一些限制。
2.4 高效pegRNA设计较为复杂
目前研究表明,PE系统中pegRNA的PBS序列、RTT序列、nicking sgRNA位置等参数均对编辑效率有较大影响[26, 31, 40-41]。因此,其设计构建较为复杂并且需要大量测试才能找到最高效的组合形式。这为如何简单、低成本地设计高效pegRNA带来了挑战。
2.5 PE系统的安全性未得到全面评估
目前已有研究表明,碱基编辑系统的脱氨酶元件异位表达会造成全基因组及转录水平的随机脱靶,为其应用带来严重的安全隐患[18-21]。由于PE引入了M-MLV RT,该酶在细胞内异位表达是否也会在全基因组或转录组水平产生随机突变,或者造成内源逆转录水平的扰动,尚未得到全面详细的评估。
此外,PE系统还存在缺少高效多基因编辑技术体系、载体过大超过腺病毒相关病毒(Adeno-associated virus,AAV)包被容量等问题,上述这些限制因素制约了PE在应用层面的发展。如何克服这一系列问题,优化PE的编辑特性,解决其瓶颈限制,对于研究学者来说是重大并且亟需解决的挑战。
3. PE系统的优化策略
基于上述存在的问题,科学家们通过多种优化改进策略对PE系统进行提升。本文对自PE系统开发以来的优化方法进行归纳(表1),将其分成下述几类。
表 1 PE的优化策略Table 1. Optimization strategies of PE种类
TypePE版本
PE version测试物种
Test species优化策略
Optimization strategy优化效率1)
Optimization efficiency参考文献
Renference植物
PlantMS2PE 水稻 原位招募RT 1.2~10.1倍 [47] PE-P3-RT-M 水稻、玉米 Cas9的N端融合RT/RTT,引入同义突变 7.0~10.0倍 [51] Pol II-PE3/PE3b 玉米、水稻 增加pegRNA转录 1.2~2.9倍 [52] PPE3-evopreQ1 水稻 epegRNA策略/高温处理 20.0%~60.5% [53] ePPE 水稻 删除RT的RNaseH结构域/添加病毒核衣壳蛋白 平均5.8倍 [54] ePPEplus/CMPE 小麦 ePPE基础上融合RT增效突变/PEmax策略 平均33.0倍 [55] PE2 (v2) 水稻 引入T5核酸外切酶 1.7~2.9倍 [56] enpPE2 水稻 Pol II-PE策略/epegRNA策略/PEmax策略 平均43.5倍 [57] PBS Tm + dual-pegRNA 水稻 设计优化PBS Tm/双pegRNA策略 2.9~17.4倍 [58] ePE2 水稻 在enpPE2基础上融合ePPE策略 1.1~1.9倍 [59] ePE5max 玉米 Pol II-PE策略/epegRNA策略/PEmax策略 1.4%~21.5% [60] 种类
TypePE版本
PE version测试物种
Test species优化策略
Optimization strategy优化效率1)
Optimization efficiency参考文献
Renference动物
AnimalepegRNA 人类 添加结构化RNA基序 3.0~4.0倍 [42] G-PE 人类 添加G四联体结构 1.7~1.9倍 [43] ePE 人类、小鼠 添加Csy4识别位点 1.9~4.9倍 [44] xr-PE 人类、小鼠 添加xrRNA结状三级结构 2.5~4.5倍 [45] sPEs/tPEs/SnPEs 人类 pegRNA结构改造 2.0~4.0倍 [49] spegRNA/apegRNA 人类 RTT引入同义突变 平均353.0倍 [50] p2PE3 人类 采用Pol II型启动子 1.6~13.3倍 [61] PE+CPC/HDACi 人类、猪 添加小分子药剂(CPC/HDACi) >4.0倍 [62] PE6 人类、小鼠 更换紧凑型RT酶/连续定向进化Cas9 24.0倍 [63] PE2* 人类 优化核定位信号序列 1.5~1.9倍 [64] PE5max 人类、小鼠 抑制DNA错配修复(MMR)/PE载体优化/引入nCas9增效突变 2.0~7.7倍 [65] hyPE2 人类 连接处添加结合蛋白Rad51结构域 1.0~2.6倍 [66] CMP-PE3 + dsgRNA 人类、小鼠 使用dead sgRNA/融合染色质调节肽 3.6~5.1倍 [67] IN-PE2 人类、小鼠 PE蛋白N端融合多肽序列 1.6倍 [68] HOPE 人类 使用双pegRNA 1.5~3.5倍 [69] 1)除ePE2的优化效率是与enpPE2相比外,其余PE版本的优化效率均是与常规PE系统比较
1) In addition to the optimization efficiency of ePE2 comparing with enpPE2, the optimization efficiency of other PE versions is compared with conventional PE system3.1 pegRNA结构及表达的优化
由于pegRNA的3'端为非结构化的PBS序列,在细胞中非常容易被消化降解,而PBS序列缺失将无法有效启动逆转录过程,进而极大影响PE的效率[42]。因此,很多研究通过向pegRNA的3'端添加特殊的RNA结构,增加其稳定性。例如Nelson等[42]将结构化RNA基序整合到pegRNA的3'端,由此产生的工程化pegRNA(epegRNA)可将多种不同细胞的编辑效率提高3.0~4.0倍。Li等[43]通过将G四联体结构引入pegRNA,同样可以显著提升PE效率。Liu等[44]利用Csy4识别位点可形成发夹结构的特点,将其融合到pegRNA的3'端提升PE的效率,并认为该方法可以减少pegRNA自身环化的现象。Zhang等[45]则是通过添加xrRNA非编码结状三级结构提升pegRNA抵抗外切酶消化的能力。Perroud等[46]通过引入植物病毒来源的TYMVtls,在小立碗藓中实现PE系统效率提升。Chai等[47]通过附加MS2适配体,同时利用MS2适配体与MCP蛋白互作的方法,实现MCP-RT融合蛋白的原位招募,将PE效率提升1.2~10.1倍。此外,Liu等[48]通过将pegRNA的PBS+RTT序列与sgRNA拆分开,形成petRNA,也可以实现与常规系统相当的效率,这个方法可能对保护pegRNA末端、减少PBS和RTT被消化起到一定作用。Feng等[49]也开发了tethered PEs (tPEs)方法,通过在pegRNA末端添加RNA适配体,并在nCas9上融合RNA适配体的识别蛋白,将pegRNA的3'末端拴住;此外,Feng等[49]也将PBS+RTT序列从pegRNA中拆分,融合RNA适配体序列并环化,开发了split pegRNA PEs (SnPEs)方法以实现精准编辑。
此外,由于pegRNA的PBS序列与靶向目标的spacer序列存在反向互补,影响PE的效率[34],为了减少其自身互补配对形成错误二级结构,Li等[50]通过在pegRNA逆转录模板的适当位置引入同义突变来开发spegRNA,使PE的效率大幅度提升。Xu等[51]也在植物中通过pegRNA引入同义突变获得了类似的结果,猜测该过程可能通过错配修复途径提升效率。
除此之外,对pegRNA的表达策略进行优化也可以显著提升PE的工作效率。多项研究均表明,采用Pol II启动子辅以RNA自剪切可以实现PE[31-32, 38-39],其中,Jiang等[52]使用的启动子为Pol II+Pol III复合型启动子,包含了35S、CmYLCV、U6的重要启动区域,在玉米中实现了高效的PE。类似的结果也在动物细胞中被证明,例如Huang等[61]、Yuan等[70]都利用Pol II启动子结合RNA自加工系统,最终实现了高效的多基因编辑。除此之外,通过对pegRNA中的sgRNA骨架结构进行优化,增加其稳定性,也可以提升PE效率[38-39, 62]。
3.2 RT效应蛋白元件的优化
RT是PE系统最核心的组成,提升RT活性或替换高活性RT是有效提升PE系统效率的方法。例如Anzalone等[26]在PE1到PE2的开发过程中向RT引入5个显著提升性能的突变,显著提升了PE效率。Lin等[31]也通过提升温度至M-MLV RT更适应的条件,在植物原生质体中将编辑效率提升1.6倍。Zou等[53]结合epegRNA策略及提升温度处理,高效地获得了水稻PE植株。Zong等[54]发现删除M-MLV RT的RNA核酸酶H(RNase H)结构域和在M-MLV RT的N端融合病毒核衣壳蛋白(Nucleocapsid,NC),可分别将其编辑效率提高2.0和3.2倍。Ni等[55]进一步在RT中引入V223A突变,可使编辑效率再提升2.8倍。Lu等[71]和Bosch等[72]通过更换效应蛋白的启动子,分别实现PE系统的编辑和传代稳定性。Perroud等[46]发现植物内源的Tnt1 RT同样具备引导编辑能力,虽然效率不及常规的基于M-MLV RT的PE系统,但更为紧凑。2023年,Doman等[63]再次升级PE系统,运用噬菌体辅助的连续进化(Phage-assisted continuous evolution,PACE)系统开发了PE6系列的全新PE,其效率更高、结构更紧凑,在基于AAV递送的体内编辑中比最先进的PE系统效率还要高24.0倍。
除了直接提升RT活性之外,对融合蛋白的结构进行优化也可以提升效率,例如Liu等[64]通过对核定位信号序列进行优化调整,提高了荧光报告系统和内源基因的编辑效率。Xu等[51]发现将RT融合在nCas9的N端,可有效提升PE系统的效率。此外,提升nCas9的识别能力,例如Chen等[65]引入nCas9的高效变体(R221K、N394K),可以有效实现PE效率的提升。该策略在植物中也有效[55]。向PE系统引入一些重要元件或结构域,也会对提升编辑能力有所帮助。例如Song等[66]在nCas9和RT结构域之间插入Rad51-DBD,产生hyPE2系统,有效提高了PE效率。Liang等[56]将T5核酸外切酶引入PE系统,其中融合了T5的PE2 (v2)版本在几个基因组位点的PE效率提高1.7~2.9倍。Park等[67]也通过融合染色质调节肽提升nCas9的识别效率,来提升PE效率。Velimirovic等[68]通过高通量筛选多肽序列,并附加于PE融合蛋白的N端,确定了一系列可以对PE效果有不同程度提升的增强肽段。Li等[57]将该方法与其他优化方法结合,成功在植物中获得了最高效率达77.08%的PE。此外,还有多个团队编辑目标基因的同时编辑抗性标记或抗除草剂基因的位点,通过抗性标记或除草剂筛选植物,从而富集编辑事件,提升PE效率[38-39]。
与PE2相比,PE3可以有效提升PE效率,但该过程需要额外引入nicking sgRNA切割编辑链的对侧链。为了简化该系统,多个团队尝试利用Cas核酸酶替代nCas9进行PE[73-75]。结果表明该方法的编辑效率与PE3相似,但副产物比例更高。这种副产物可以通过抑制NHEJ途径(例如使用小分子抑制剂AZD7648进行抑制),提升正确编辑的比例[74]。此外,Qi等[62]也发现一些小分子例如组蛋白去乙酰化酶抑制剂等也对提升PE效率有所帮助。
在递送系统上,Sürün等[76]利用化学修饰的pegRNA及nCas-RT融合蛋白的mRNA形式向人类诱导性多功能干细胞递送PE,实现了荧光蛋白报告系统的碱基置换。Petri等[77]利用RNP的方法在体外组装PE复合体,再进行递送,实现DNA-free的递送方法。此外,多个团队也通过成功截短RT、将PE载体拆分至2个AAV病毒分别进行包被等方法压缩PE,实现了基于AAV的递送[78-83]。以上方法均实现了PE系统应用场景的扩展。
3.3 靶向范围的优化
常规PE使用的nCas9来源于化脓性链球菌,其特定的“NGG”的PAM需求限制了它在全基因组的靶向范围。为此,多个研究团队尝试替换SpCas9,拓宽PE靶向范围。Hua等[84]和Aird等[85]分别在植物和动物中构建来源于金黄色葡萄球菌Staphylococcus aureus的SaCas9的PE系统,将PE的识别PAM基序拓宽至“NNGRRT”序列。Lin等[58]使用SpG变体替换SpCas9,实现了PAM为“NG”的编辑,极大扩展了PE的靶向范围尤其是dual-pegRNA的靶向范围。Kweon等[86]通过使用SpCas9-NG、SpG及SpRY变体,极大拓宽了PE系统在全基因组的可编辑范围,使PE可以靶向基因组的任意位置。此外,Oh等[87]使用来源于新凶手弗朗西丝菌Francisella novicida的FnCas9为PE的设计提供更多的选择,虽然其识别PAM基序与经典的SpCas9相同,但其切口位置更为靠前,通常为PAM序列上游的6~8 bp,而常规的SpCas9的非靶标链切口通常为上游的3~4 bp[4, 87-88]。
3.4 DNA修复通路的优化
由于PE涉及到多个复杂的修复过程,因此,寻找其中的关键步骤或通路,改变DNA修复途径,理论上将有助于PE系统的改良。基于上述理论,Chen等[65]通过CRISPRi筛选,抑制修复相关蛋白表达,同时测试PE系统的效率,发现MSH2、MSH6、MLH1和PMS2这些DNA错配修复途径(Mismatch repair,MMR)中的关键蛋白显著影响PE,并通过RNAi和MMR通路缺失的HAP1细胞试验证明抑制MMR通路可以有效提升PE效率;此外,还通过融合功能缺失型突变MLH1dn,开发了PE4和PE5,以降低MLH1对目标位点的修复识别,从而显著提升PE系统效率。Da Silva等[89]也针对32个DNA修复因子进行了遗传筛选,这些修复因子涵盖了所有已经被报道过的相关修复途径,同样发现抑制MMR途径可有效提升PE效率[65]。
3.5 高效pegRNA设计策略
Anzalone等[26]研究表明,PE系统中的PBS序列、RTT序列、nicking sgRNA位置等均对PE效率有非常大的影响,但是如何设计能够使PE效率最大化,这些参数似乎并没有非常明显的规律。因此在通常情况下,科学家需要大量测试才能找到最高效的pegRNA形式,费时费力。针对上述问题,多个科学团队开发了辅助pegRNA设计的网站,以简化pegRNA的设计流程。例如pegFinder[90]和PrimeDesign[91]这2种软件均可以根据目标位点和所需编辑设计出一系列pegRNA,并提供优先排列。其中,pegFinder可针对多种不同Cas9变体设计PE2和PE3;PrimeDesign可以针对单种类型的编辑或多种类型组合的编辑进行设计,也可以实现饱和突变的筛选;该设计团队还开发了PrimeVar,可针对已知人类疾病变异位点数据库快速选择pegRNA和nicking sgRNA。除了上述2个网站,multicrispr[92]和PnB Designer[93]工具也在针对基因敲除或单碱基编辑进行sgRNA设计的基础上增加了针对pegRNA进行设计的功能。Standage-Beier等[94]还开发了可以高通量设计pegRNA及提供对应引物的PINE-CONE。Morris等[95]开发的Prime Editing Design Tool在线网站工具,可以实现针对所需致病变异的对应pegRNA的筛选和设计。此外,Hwang等[96]开发的PE-Designer也可以实现用户友好、便捷的pegRNA设计。
Lin等[58]通过总结PBS规律,发现PBS的熔解温度与PE的效率显著相关,在水稻中PBS的熔解温度为30 ℃时PE具有高效率。类似结果也在Kim等[97]的高通量结果中得到印证,除了目标位点sgRNA识别的效率外,同一位点PBS序列的G、C碱基的比例及熔解温度也是影响PE的重要因素。为此,Lin等[58]和Jin等[98]开发了PlantPegDesigner设计网站,帮助研究人员简化高效pegRNA设计流程;试验证明,PlantPeg Designer由于有真实的植物试验数据支持,其在植物中的表现优于多数pegRNA设计网站。
由于高效pegRNA的设计规律较为复杂,科学家们尝试通过深度学习的方法寻找pegRNA设计与编辑效率的关系。Kim等[97]设计了54 836个pegRNAs进行高通量测试,对pegRNA的PBS序列、RT模板序列及靶向位点的间隔序列进行了长度、退火温度、碱基偏好等的深入分析,发现靶向位点的Cas9核酸酶活性,PBS序列的G、C碱基的比例,熔解温度,PBS及RT模板序列上2个U碱基的个数等,都是影响PE系统效率的重要因素;他们还通过机器学习开发了预测PE效率的方法计算模型DeepPE,并发现该模型计算结果与试验结果相拟合。Koeppel等[99]也设计了3 604种不同的pegRNA序列,用于实现不同类型的片段插入,他们在3种细胞的4个不同位点进行高通量试验,发现在不同PE和不同DNA修复环境下,插入序列的长度、核苷酸组成和二级结构都会影响插入效率,并且TREX1和TREX2会对大片段插入造成一定影响;他们还开发了机器学习模型预测效率,并能和试验数据有较好的相关性。此外,Li等[100]也利用多个已发表的数据结果进行训练,开发了基于机器学习的程序Easy-Prime,为海量的GWAS变异提供了高效的pegRNA设计。
为了进一步提升PE系统的效率,多个科学团队均尝试了用成对的pegRNA策略进行编辑。所谓成对的pegRNA,指的是针对目标位点同时在正义链和反义链上设计pegRNA。2个pegRNA的PBS+RT序列间存在反向互补序列,利用他们之间的互补配对,可能有利于将所需编辑引入基因组中。Lin等[58]在植物中进行测试,提出了dual-pegRNA策略,即针对正义链和反义链同时设计pegRNA实现相同编辑,该策略在15个内源位点的测试结果表明,dual-pegRNA的策略在多数内源位点上的效率均不低于单个pegRNA策略,平均效率可以提高3.0倍;他们也将该策略整合至PlantPegDesigner设计网站。Zhuang等[69]开发了基于成对pegRNA的HOPE系统,在293T细胞及HCT116细胞中都获得了更为高效的编辑;与常规的PE3系统相比,HOPE系统的副产物更少,可以获得更高的产物纯度。
3.6 优化大片段操纵能力
大片段操纵是PE系统需要克服的重大瓶颈。为了克服以上问题,多个团队通过结合特异性重组酶的方法来实现大片段定点插入。Yarnall等[101]开发了PASTE方法,使用单个pegRNA插入位点特异性重组酶的识别序列,利用重组酶的定点整合实现大片段插入,在3种不同细胞系的多个位点中,实现了最高36 kb的大片段定点插入;他们还通过宏基因组挖掘和工程化改造同源物确定了25 614种位点特异性丝氨酸重组酶及其DNA识别位点,大大扩展了位点特异性丝氨酸重组酶的多样性。此外,运用成对pegRNA的方法已成功应用于高效的定点大片段插入。例如Wang等[102]基于成对的pegRNA开发了GRAND editing新方法,所需插入的大片段信息被装载在pegRNA的RTT序列上,2个pegRNA的RTT序列之间存在互补序列,但2个RTT序列均与基因组序列不相似;运用该方法,他们实现了为20 bp~1 kb的精准插入,包括效率高达63.0%的150 bp的插入和28.4%的250 bp的插入。Li等[59]也运用了类似的策略,结合PE的其他优化方法,实现植物基因组中HIS、HA、FLAG等蛋白标签的有效插入。设计成对的pegRNA插入重组酶识别位点,并结合位点特异性丝氨酸重组酶的方式,也可以实现高效率的大片段定点插入。例如Anzalone等[103]开发的twinPE系统,能够在人类细胞中定点整合>5 000 bp的大片段和实现40 kb的DNA倒位。2023年,Sun等[104]开发了PrimeRoot编辑工具,在植物中建立了基于PE的大片段定点插入新策略,该策略运用优化的dual-pegRNA插入重组酶识别位点,结合优化的位点特异性重组酶,实现了高达11.1 kb的定点大片段DNA插入。
成对的pegRNA策略同样也用于大片段的精准删除。例如Choi等[105]开发了PRIME-Del的DNA大片段删除技术,他们利用成对pegRNA靶向基因组待删除位置,并将这2个pegRNA的RTT序列设计为分别与另一pegRNA产生的断裂处的基因组序列匹配;这种方法的精准删除比例比基于Cas9和成对的sgRNA产生的大片段精准删除的比例更高。Jiang等[106]通过构建基于Cas9核酸酶的PE,利用成对pegRNA间互补配对实现小片段序列的插入并替换原基因组中的DNA大片段,实现插入不超过60 bp的情况下直接删除1~10 kb的基因组大片段,并成功在酪氨酸血症小鼠模型中修复了1个由1. 38 kb大片段产生的致病性位点。Tao等[75, 107]也通过成对的pegRNA和Cas9核酸酶相结合,相继开发了Bi-PE和WT-PE新策略,实现高效的大片段删除。其中WT-PE成功创制了高达16.8 Mb的大片段删除的案例,为大片段畸变导致的疾病的模型创制和疾病治疗提供了新的方法。
除了大片段插入及删除,Kweon等[108]也运用基于成对pegRNA的PE实现了基因组的易位和倒位,Tao等[107]也通过WT-PE实现了染色体易位。
3.7 同时针对多基因进行PE
多基因编辑对于重要性状聚合等均有重要的意义。目前,有多个团队测试了基于PE系统进行多基因编辑的方法。Jiang等[52]将多个pegRNA分别转录,在玉米中实现了ALS基因(W542L和S621I)的同时突变。Yuan等[70]利用tRNA具有启动子和RNA加工的双功能,在不同pegRNA序列间加入间隔序列,开发了紧凑的hCtRNA-M阵列,实现了3个位点同时编辑,多基因编辑的最高效率可达57.5%。Huang等[61]也运用Pol II启动子转录含有poly(T)序列的pegRNA,通过Csy4系统对多个pegRNA进行加工,实现了多个基因的同时编辑。Li等[109]等运用上述提到的富集编辑策略,并运用tRNA对多个pegRNA的转录本进行加工,实现了多个内源基因的同时、精确的编辑。Ni等[55]通过开发增强版本的ePPEplus,结合Csy4策略,在小麦原生质体中实现了10个基因的同时编辑,并在小麦植株中同时编辑8个基因位点,极大地扩展了PE在实现农艺性状叠加方面的适用性。
3.8 特异性评估
基因组编辑工具的脱靶效应,根据其作用原理通常可分为两类:一类为传统意义上的sgRNA(或pegRNA)依赖型的脱靶效应,这是由于脱靶位点序列与sgRNA的识别序列具有较高的相似性,使Cas9与sgRNA形成的复合体错误地结合到脱靶位点所致;另一类为sgRNA(或pegRNA)非依赖型的脱靶效应,通常情况下是由于基因编辑工具的组成元件异位表达所致。例如碱基编辑系统中脱氨酶的异位表达,会引起全基因组或转录水平的随机脱氨事件,造成难以预测的由脱氨酶引起的脱靶[18-21]。与其他常规的CRISPR/Cas基因编辑系统相比,PE系统需要引入RT这一元件,其异位表达是否也会造成全基因组或者转录本难以预测的随机脱靶事件,需要详细、全面的评估。当然,PE系统的pegRNA依赖型脱靶水平也需要深入研究。Anzalone等[26]在细胞系中的测试结果表明,PE系统的pegRNA依赖型脱靶频率很低,猜测主要是由于PE除了需要满足sgRNA与靶向序列之间的识别之外,还需要满足PBS与非靶向链的识别,以及逆转录产生的flap结构整合至基因组时与基因组序列的识别;此外,他们也在细胞系层面证明PE在转录水平上不会造成基因表达量的显著变化。Kim等[110]通过改进的nDigenome-seq技术对PE的脱靶效应进行了全面的分析,也证明PE系统的pegRNA依赖型的脱靶频率较CRISPR/Cas9更低,并且可以通过使用Cas9的高保真变体,在保证一定程度的编辑效率的情况下,进一步提升PE系统的保真性。Jin等[111]也利用原生质体脱靶位点的深度靶向测序与植物个体水平全基因组测序结合的方法检测了PE系统pegRNA依赖型的脱靶效应,发现其脱靶频率较低,且受错配的数量和位置的影响,可以通过理性设计pegRNA减少脱靶。综上所述,PE鲜有pegRNA依赖型脱靶事件的发生。
此外,Jin等[111]还以水稻为代表在个体水平上进行了PE系统的pegRNA非依赖型脱靶的评估,全基因组测序没有检测到PE系统在全基因组范围内额外产生的碱基变换和增删突变。Jin等[111]还分析了逆转录转座子OsTos17和端粒区域的拷贝数及保真性,发现PE系统中RT的异位表达不影响内源逆转录转座子和端粒酶的活性。此外,PE也不会对内源mRNA造成逆转录及基因组整合。Schene等[112]也通过对基因组编辑后的细胞团进行全基因组测序,分析了PE在全基因组范围内的特异性,获得了类似的结果。Gao等[113]构建背景突变数量极低的细胞系,敏锐地分析脱靶突变事件。全基因组和转录组范围分析结果显示PE3没有产生额外的pegRNA非依赖型的脱靶事件。Habib等[114]对人类多能干细胞进行评估的结果也表明PE的RT结构域的引入不会导致基因组发生随机突变。因此,PE系统不会造成pegRNA非依赖型脱靶事件的发生,总体来说,PE系统特异性较高,暂时没有发现安全性上的隐患。
4. PE系统的应用
与CRISPR/Cas9编辑技术和碱基编辑技术产生的编辑类型相比,PE系统突破了编辑限制,可以实现几乎任意形式的编辑类型。因此,该系统自开发以来就受到了众多科研工作者的高度关注和青睐,并迅速应用于细菌(如大肠埃希菌),动物(如小鼠、果蝇、斑马鱼、兔子、猪等),植物(如水稻、玉米、番茄、小麦、烟草、小立碗藓、马铃薯、拟南芥等)及各种人类细胞系中。本综述简略按照应用系统的区别对PE系统在生物系统中的应用现状进行总结。
4.1 PE系统在植物育种中的应用
基因编辑技术对农业发展产生了深刻的影响,其应用已经非常广泛。2023年4月28日,农业农村部发布《2023年农业用基因编辑生物安全证书(生产应用)批准清单》,下发了全国首个植物基因编辑安全证书,体现了国家对作物基因组编辑育种产业化的战略布局。基因编辑技术在农业上积累了丰富的研发及应用经验,正在逐渐转变为一项常规的农业生产技术,参与重塑未来农业的行业生态。PE系统作为基因组编辑育种中编辑性能最优越的代表,尽管开发至今未满4年,却以极快的速度在新型种质资源创制等重要农艺性状的开发上得以应用。PE技术开发之初,就有多个研究小组在多种重要作物中进行测试,如水稻、小麦、玉米、番茄等[31, 52, 71]。该系统可以实现碱基任意互换,为创制功能获得性突变(Gain-of-function mutation)提供了高效便捷的方法,最常见的应用方向是抗除草剂种质的创制[37, 115]。多个团队利用PE创制了常规碱基编辑器难以获得的新抗除草剂基因突变类型。例如Jiang等[116]通过使用优化的PE系统,在水稻抗草甘膦基因(EPSPS)中有效地产生了可遗传的TAP-IVS突变,克服了之前抗草甘膦突变类型无法获得纯合后代的瓶颈问题,为水稻的非转基因草甘膦抗性育种提供了全新的技术路径。Xu等[117]运用PE系统针对抗除草剂基因OsACC1的重要位点进行饱和式筛选,鉴定出一系列赋予除草剂耐受性的全新突变类型,并将其应用到水稻育种中。Qiao等[60]也采用多种PE优化方法,成功产生了可遗传的抗除草剂玉米突变株系。Gupta等[118]通过PE系统赋予水稻白叶枯病抗性。此外,Zhang等[119]也运用PE系统创制功能获得性突变,赋予了烟草高水平的顺−冷杉醇合成能力。
4.2 PE系统在动物及疾病治疗中的应用
据OMIM数据库(人类孟德尔病数据库)的统计,目前已知的人类单基因遗传病有7 000多种,其中4 000多种的致病基因和发病机制比较明确,而多数遗传病的治疗需求仍远远未被满足。PE系统能够对致病基因的序列直接进行修改,纠正异常基因,从而达到治疗疾病的目的,具有一次治疗终身治愈的潜力。目前PE已经在多种不同类型的人类细胞或组织中实现了编辑,例如Chemello等[120]利用PE系统在人类心肌细胞中对DMD基因的第52号外显子进行编辑,成功恢复了细胞内抗肌萎缩蛋白的表达;Sun等[121]对雄激素受体突变的iPSCs细胞进行修复,使其恢复分化为3个胚层的能力。这些结果表明,PE系统在人类疾病治疗中有良好的应用前景。Schene等[112]利用PE3系统在功能上成功纠正了肠道类器官中DGAT1基因的致病突变和肝脏类器官中ATP7B基因的致病突变。
此外,PE系统可以应用在动物中进行重要疾病模型的创制以研究其致病机理。PE还可以在动物遗传育种中发挥重要作用,创制畜禽重要优异性状,也可以创制重要突变类型为基础研究提供技术支持。在非哺乳动物模型上,Bosch等[72]在果蝇S2R细胞中测试了引导编辑器的效率,他们利用引导编辑器将终止密码子引入3个标记基因中,编辑效率分别为35.2%、11.6%和21.9%,并进一步在黑腹果蝇中实现了精准编辑。Petri等[77]也将PE系统应用到斑马鱼胚胎中,成功诱导了可遗传突变,突变率高达30%。在哺乳动物模型上,PE也有很多应用成功的例子,例如Liu等[64]基于双AAV将PE分成两部分递送到成年小鼠体内,成功纠正小鼠肝脏中的致病性突变。Qian等[122]将PE系统成功地应用到兔子身上,并使用PE系统产生了一个新的TSD(一类性神经退行性疾病)兔模型。Qi等[62]证明PE系统也可以编辑猪的基因组,拓宽了PE系统的研究及应用范围。最近,Davis等[79]基于优化双AAV的引导编辑器递送系统,在小鼠多个重要器官包括大脑(皮层中编辑效率高达42%)、肝脏(编辑效率高达46%)和心脏(编辑效率高达11%)中都实现了精准编辑。以上研究充分展示了PE系统在体内实现基因遗传疾病治疗的应用前景。
5. PE系统发展前景
PE系统以CRISPR/Cas技术为基础,实现了基因组中灵活的精准编辑。针对其进行的开发及优化,大大提升了编辑效率和对大片段的操纵能力。尽管如此,目前整体上PE的编辑效率与碱基编辑技术等与其他方法相比仍有差距,尤其PE在不同物种、不同细胞类型、不同靶向位点中的差异仍然很大,这导致PE系统在很多情况下仍然比较吃力。因此,进一步优化提升PE的编辑能力和DNA大片段操纵能力仍然十分重要。目前,PE系统的优化仍不断有新突破,尤其是最新出现的PE6系列优化版本,通过PACE进化系统对PE系统中的Cas9组分进行定向进化,使PE效率得以显著提升[63]。结合最新出现的针对RT进行优化的ePPEplus[55]等策略,相信可以极大地拓宽PE的编辑场景。
目前PE系统相关的研究主要集中于编辑特定的目的靶点及基因组内的位点,对高通量筛选、基因组外遗传物质的编辑(如线粒体、叶绿体基因组)等场景的研究还不充分;此外,虽然目前PE系统未发现全基因组或转录水平的脱靶效应,但随着其编辑能力的进一步提升,该方法的脱靶风险仍需谨慎的评估;PE系统本身还存在载体较大的问题。因此,如何开发高效、安全的递送系统,保证PE系统在动植物体内特定部位(或者组织类型)的有效表达,也是其应用的关键因素。综上所述,PE系统为疾病治疗、农业育种、基础研究等领域提供了高效、安全、灵活的基因编辑平台,通过未来的优化改进,PE系统将为上述领域提供更为稳健的技术支撑。
致谢:感谢河北农业大学生命科学学院李君教授在文章写作方面给予的建议和帮助。
-
![]()
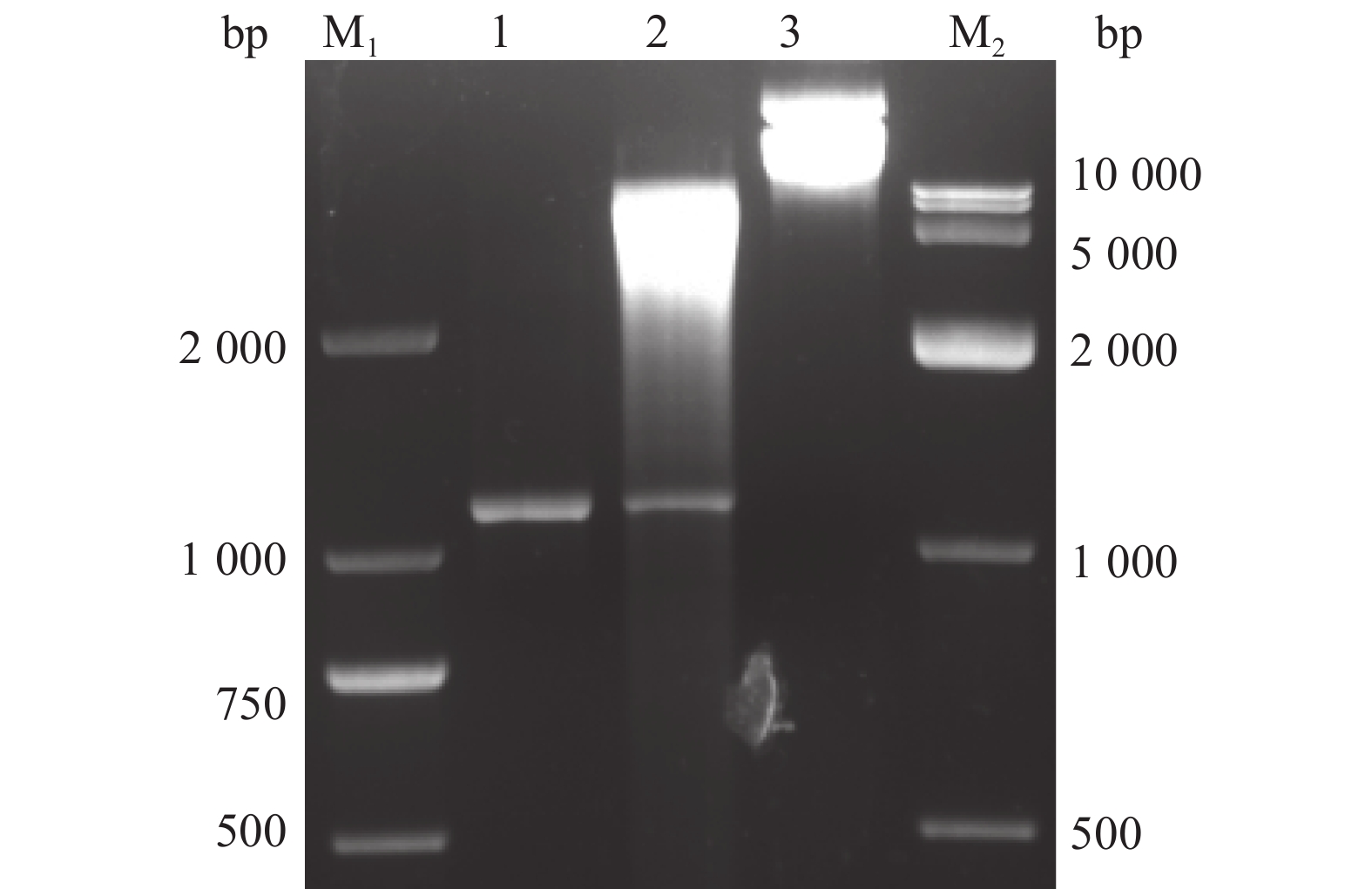
图 1 重组质粒pET-32a-Cp51酶切鉴定
M1:DNA Marker DL2000;1:Cp51目的基因;2:pET-32a-Cp51双酶切;3:pET-32a-Cp51阳性质粒;M2:DNA Marker DL10000。
Figure 1. Identification of recombinant plasmid pET-32a-Cp51 by enzyme digestion
![]()
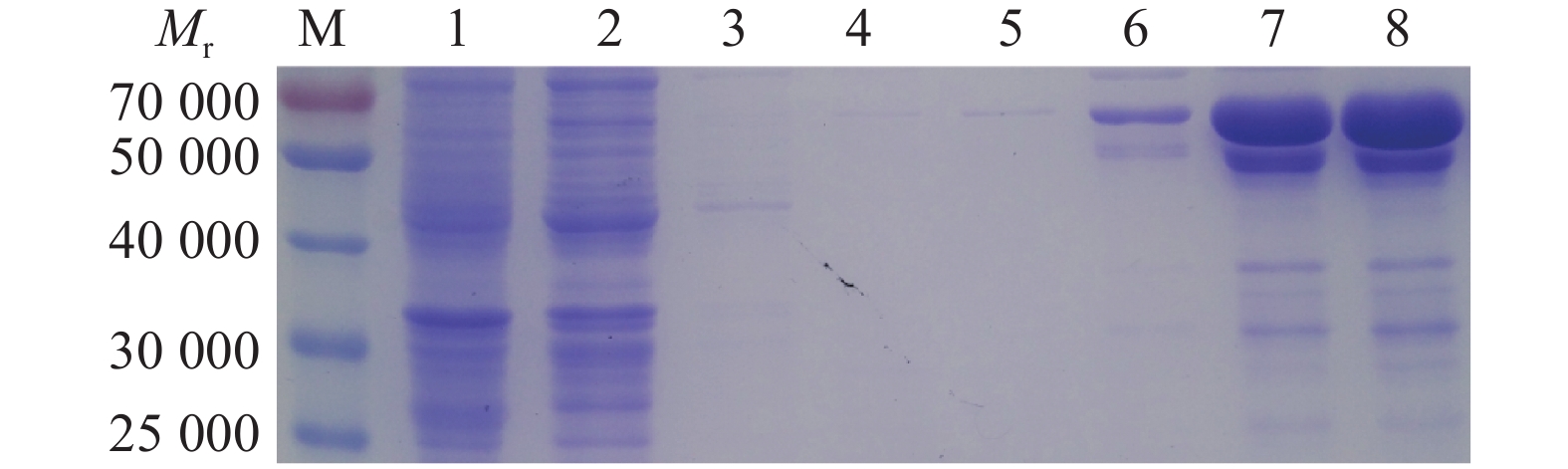
图 2 pET-32a-CP51表达SDS-PAGE电泳
M:蛋白Marker;1:pET-32a(+)未诱导;2:pET-32a(+)诱导;3:pET-32a-Cp51未诱导;4:pET-32a-Cp51诱导上清;5:pET-32a-Cp51诱导沉淀。
Figure 2. SDS-PAGE pattern of expressed pET-32a-CP51 protein
![]()
图 3 pET-32a-Cp51蛋白纯化的SDS-PAGE电泳
M:蛋白Marker;1:pET-32a(+);2蛋白流穿液;3:洗涤液;4:30 mmol·L–1咪唑洗脱液;5:40 mmol·L–1咪唑洗脱液;6:100 mmol·L–1咪唑洗脱液;7:150 mmol·L–1咪唑洗脱液;8:200 mmol·L–1咪唑洗脱液。
Figure 3. SDS-PAGE pattern of purified pET-32a-Cp51 protein
![]()
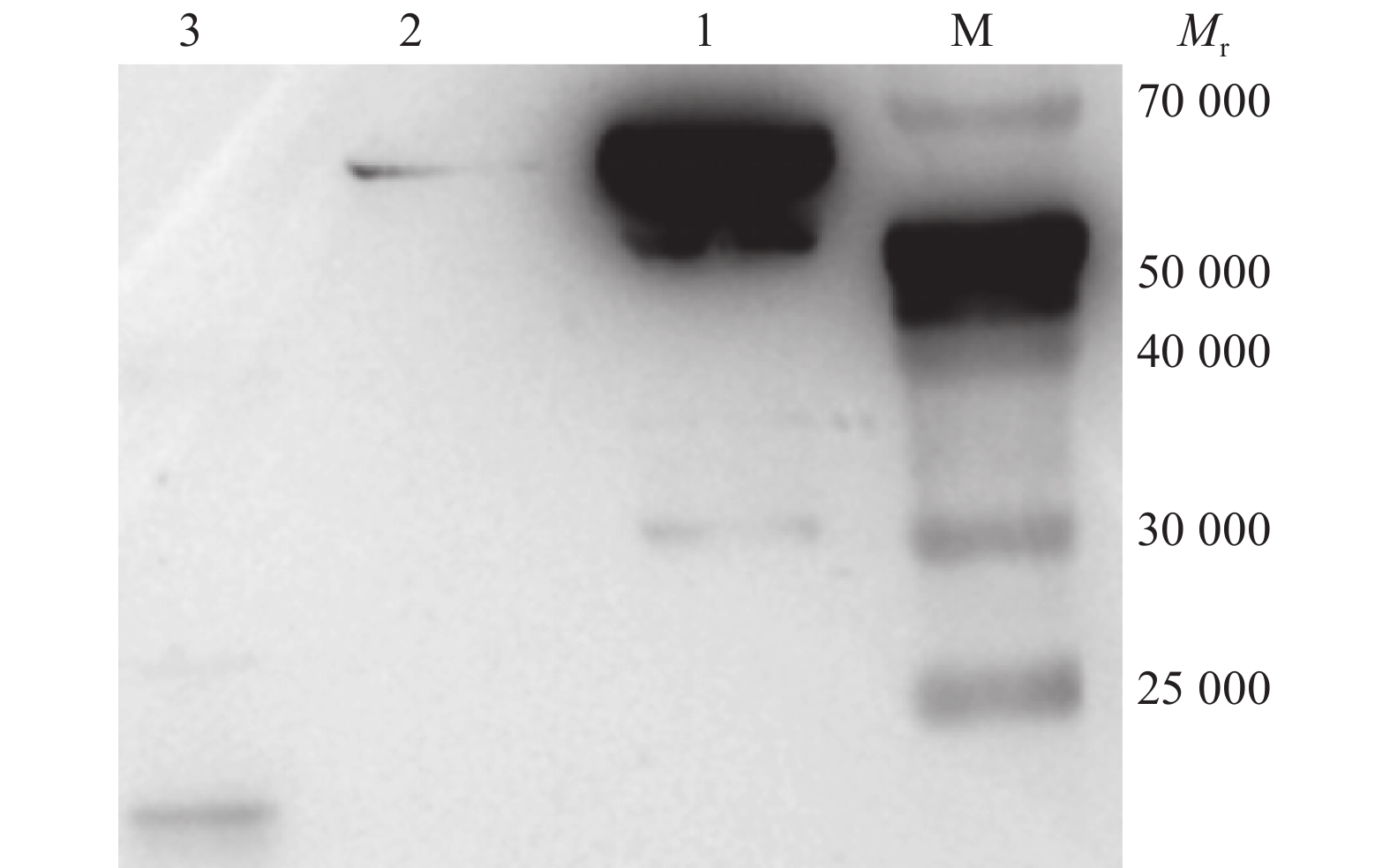
图 4 pET-32a-Cp51蛋白的Western blot检测结果
M:蛋白Marker;1:pET-32a-Cp51 150 mmol·L–1咪唑洗脱液;2:pET-32a-Cp51未诱导;3:pET-32a(+)诱导。
Figure 4. Western blot results of pET-32a-Cp51 protein
![]()
图 5 不同质量浓度pET-32a-Cp51裂解酶对A型产气荚膜梭菌的活性
Figure 5. Activity of different concentrations of pET-32a-Cp51 lysin against type A Clostridium perfringens
![]()
图 6 pET-32a-Cp51裂解酶对A型产气荚膜梭菌不同分离株的活性
Cpxxc为广东新兴鸡样品产气荚膜梭菌分离株,Cpxxd为广东新兴鸭样品产气荚膜梭菌分离株。
Figure 6. Activity of pET-32a-Cp51 lysin against different isolates of type A Clostridium perfringens
-
[1] GARCIA J P, BEINGESSER J, FISHER D J, et al. The effect of Clostridium perfringens type C strain CN3685 and its isogenic beta toxin null mutant in goats[J]. Vet Microbiol, 2012, 157(3/4): 412-419.
[2] LOVLAND A, KALDHUSDAL M. Severely impaired production performance in broiler flocks with high incidence of Clostridium perfringens-associated hepatitis[J]. Avian Pathology, 2001, 30(1): 73-81.
[3] 汪复, 朱德妹, 胡付品, 等. 2012年中国CHINET细菌耐药性监测[J]. 中国感染与化疗杂志, 2013, 13(5): 321-330. [4] FILE T M, Jr, SRINIVASAN A, BARTLETT J G. Antimicrobial stewardship: Importance for patient and public health[J]. Clin Infect Dis, 2014, 59(S3): S93-S96.
[5] FISCHETTI V A. Bacteriophage endolysins: A novel anti-infective to control gram-positive pathogens[J]. Int J Med Microbiol, 2010, 300(6): 357-362.
[6] SCHRAG S J, MCGEE L, WHITNEY C G, et al. Emergence of Streptococcus pneumoniae with very-high-level resistance to penicillin[J]. Antimicrob Agents Chemother, 2004, 48(8): 3016-3023.
[7] SCHUCH R, PELZEK A J, RAZ A, et al. Use of a bacteriophage lysin to identify a novel target for antimicrobial development[J]. PLoS One, 2013, 8(4): e60754.
[8] SCHUCH R, NELSON D, FISCHETTI V A. A bacteriolytic agent that detects and kills Bacillus anthracis[J]. Nature, 2002, 418(6900): 884-889.
[9] LOESSNER M J. Bacteriophage endolysins: Current state of research and applications[J]. Curr Opin Microbiol, 2005, 8(4): 480-487.
[10] 李跃, 宫鹏娟, 夏斐斐, 等. 金黄色葡萄球菌噬菌体裂解酶LysK的表达及其多克隆抗体的制备[J]. 中国兽医学报, 2014, 34(1): 45-49. [11] 王彬. 金黄色葡萄球菌噬菌体裂解酶LysGH15外用制剂的初步研制[D]. 长春: 吉林大学, 2015. [12] 陈蔚青, 王晓枫, 王普, 等. 链球菌噬菌体裂解酶在大肠杆菌中的表达、纯化及活性检测[J]. 生物工程学报, 2009(8): 1267-1272. [13] 吴蒙, 陆海荣, 黄青山. 金黄色葡萄球菌噬菌体裂解酶Ply187的CHAP结构域的表达及抗菌活性分析[J]. 生物技术通报, 2016, 32(9): 232-238. [14] GERVASI T, HORN N, WEGMANN U, et al. Expression and delivery of an endolysin to combat Clostridium perfringens[J]. Appl Microbiol Biotechnol, 2014, 98(6): 2495-2505.
[15] MARTIN H, WILLEY B, LOW D E, et al. Characterization of Clostridium difficile strains isolated from patients in Ontario, Canada, from 2004 to 2006[J]. J Clin Microbiol, 2008, 46(9): 2999-3004.
[16] LAWRENCE R, JEYAKUMAR E. Antimicrobial resistance: A cause for global concern[J]. BMC Proc, 2013, 7(S3): S1.
[17] BORYSOWSKI J, WEBER-DABROWSKA B, GÓRSKI A. Bacteriophage endolysins as a novel class of antibacterial agents[J]. Exp Biol Med (Maywood), 2006, 231(4): 366-377.
[18] GU J, XU W, LEI L, et al. LysGH15, a novel bacteriophage lysin, protects a murine bacteremia model efficiently against lethal methicillin-resistant Staphylococcus aureus infection[J]. J Clin Microbiol, 2011, 49(1): 111-117.
[19] SCHMELCHER M, POWELL A M, BECKER S C, et al. Chimeric phage lysins act synergistically with lysostaphin to kill mastitis-causing Staphylococcus aureus in murine mammary glands[J]. Appl Environ Microbiol, 2012, 78(7): 2297-2305.
[20] PASTAGIA M, EULER C, CHAHALES P, et al. A novel chimeric lysin shows superiority to mupirocin for skin decolonization of methicillin-resistant and -sensitive Staphylococcus aureus strains[J]. Antimicrob Agents Chemother, 2011, 55(2): 738-744.
-
期刊类型引用(2)
1. 李福权,张瑞祥,赵哲,陈乐天,谢先荣. 基因编辑设计和分析辅助工具的研究进展. 科学通报. 2025(16): 2449-2467 .  百度学术
百度学术
2. 王晨雨,刘孟军,王立新,刘志国. CRISPR/Cas9技术研究进展及其在园艺植物中的应用进展. 园艺学报. 2024(07): 1439-1454 . 百度学术
其他类型引用(3)

 下载:
下载:

计量
- 文章访问数: 1478
- HTML全文浏览量: 16
- PDF下载量: 1581
- 被引次数: 5
